UNIVERSITÀ DEGLI STUDI DELLA BASILICATA Facoltà di Agraria CORSO DI LAUREA IN SCIENZE E TECNOLOGIE ALIMENTARI TESI SPERIMENTALE Sviluppo di un metodo cromatografico per la determinazione del lattulosio nel latte trattato termicamente mediante HPAEC-PAD RELATORE: Prof. Tommaso R. I. CATALDI CORRELATORE: Prof. Sabino A. BUFO LAUREANDO: Massimiliano ANGELOTTI ANNO ACCADEMICO 1997-1998
Transcript
UNIVERSITÀ DEGLI STUDI DELLA BASILICATA
Facoltà di Agraria
CORSO DI LAUREA IN SCIENZE E TECNOLOGIE ALIMENTARI
TESI SPERIMENTALE
Sviluppo di un metodo cromatografico per la determinazione del
lattulosio nel latte trattato termicamente mediante HPAEC-PAD
RELATORE:
Prof. Tommaso R. I. CATALDI
CORRELATORE:
Prof. Sabino A. BUFO
LAUREANDO:
Massimiliano ANGELOTTI
ANNO ACCADEMICO 1997-1998
INDICE
I
INDICE
1. INTRODUZIONE
1.1. Produzione e trattamento del latte di qualità pag. 1
1.2. Effetti del calore sulla composizione del latte pag. 3
1.3. Il lattulosio pag. 5
1.4. Esame dei metodi classici per la determinazione del lattulosio pag. 7
1.5. Scopo della tesi: sviluppo di un nuovo metodo cromatografico
per la determinazione del lattulosio pag. 15
2. MATERIALI E METODI
2.1. Reagenti pag. 18
2.2. Preparazione del campione pag. 20
2.3. Strumentazione pag. 22
3. TECNICHE SPERIMENTALI
3.1. Cromatografia pag. 24
3.2. Parametri cromatografici pag. 25
3.3. Cromatografia liquida ad alte prestazioni pag. 28
3.4. Rivelazione amperometrica pulsata pag. 32
INDICE
II
4. RISULTATI E DISCUSSIONE
4.1. Fase mobile: concentrazione dell’NaOH pag. 35
4.2. Stabilità dei tempi di ritenzione: effetto del carbonato pag. 41
4.3. Strategie per migliorare la riproducibilità dei tempi di ritenzione pag. 43
4.4. Effetto dei cationi sulla rivelazione amperometrica pulsata pag. 50
4.5. Analisi di un campione reale: trattamento del campione di latte pag. 52
4.6. Analisi di un campione reale: problemi legati alle concentrazioni
relative di lattosio e lattulosio presente nei campioni di latte pag. 56
4.7. Strategie di miglioramento dei parametri cromatografici pag. 59
4.8. Analisi quantitativa pag. 74
4.9. Campioni reali pag. 78
4.10. Conclusioni pag. 90
Bibliografia pag. 92
1-INTRODUZIONE
1
1. INTRODUZIONE
1.1. PRODUZIONE E TRATTAMENTO DEL LATTE DI QUALITÀ
Negli ultimi anni vi è stata una notevole diffusione di prodotti alimentari di
“alta qualità”, i quali sono sottoposti a trattamenti industriali tali da preservarne le
caratteristiche nutrizionali ed organolettiche proprie del prodotto “fresco”. Il settore
del latte alimentare è regolato da una serie di normative che sottolineano
l’importanza della qualità del latte ed il suo valore nutritivo in ciascuna delle
differenti tipologie messe in commercio. Il DPR 54/97, che recepisce la normativa
CEE 92/46, stabilisce quali devono essere le norme di produzione e qualità del
prodotto, senza peraltro definire quali devono essere i parametri da utilizzare come
indici di qualità.
Il trattamento di risanamento del latte che viene usualmente applicato è quello
termico, il quale assicura la sanitizzazione del prodotto e permette la sua conservazione
per un periodo più o meno lungo. Il latte è il prodotto secreto dalla ghiandola
mammaria delle femmine dei mammiferi. Con la denominazione latte si intende
generalmente quello di vacca, altrimenti deve esserne indicata la specie animale di
provenienza. Dal punto di vista chimico-fisico è un’emulsione di grasso in acqua
contenente zuccheri, sali minerali e proteine. Una tale composizione rende il latte un
alimento completo ad alto valore nutrizionale. È tuttavia altamente deperibile e quindi
deve subire opportuni trattamenti di conservazione. È commercializzato come latte
intero (grasso non inferiore al 3,2%), parzialmente scremato (grasso 1,5 ÷ 2,8%) e
1-INTRODUZIONE
2
scremato (grasso inferiore allo 0,3%). La frazione glucidica è rappresentata in forma
preponderante dal lattosio (circa 4,8% p/v). Si è diffuso inoltre un tipo di latte,
destinato all’alimentazione di soggetti intolleranti al lattosio, ricco in galattosio e
glucosio formatisi per idrolisi enzimatica del disaccaride. Le proteine (3,5%) sono tra
quelle con più alto valore biologico. Infine il latte contiene le vitamine A, D, E e K a
concentrazione molto diversa [1].
Le tecniche di conservazione differiscono in relazione al tipo di latte
commercializzato. La pastorizzazione è applicata al latte fresco e distrugge la
maggior parte dei microrganismi presenti. La tecnica attualmente utilizzata è il
riscaldamento del latte per 15 ÷ 40 secondi a 72 ÷ 75 °C e successivo raffreddamento
a 2 ÷ 4 °C; il prodotto che si ottiene, latte fresco pastorizzato, può essere conservato
(shelf-life) per 3 ÷ 4 giorni se mantenuto a 2 ÷ 4 °C.
Con la sterilizzazione si ottiene un prodotto stabile per un tempo più lungo
definito latte UHT. In questo caso il latte è trattato a temperature di 140 ÷ 150 °C per
pochi secondi, subito raffreddato e confezionato asetticamente (shelf-life 90 giorni a
temperatura ambiente). Il latte sterilizzato è prima confezionato, generalmente in
bottiglie di vetro, e poi sottoposto a trattamento in autoclave a 120 °C per circa 30
minuti (shelf-life 180 giorni a temperatura ambiente). Altri tipi di latte a lunga
conservazione sono: il latte condensato, concentrato per evaporazione sotto vuoto del
50% dell’acqua contenuta, previa aggiunta di sciroppo di zucchero, ed il latte in
polvere in cui l’acqua è stata allontanata quasi del tutto.
1-INTRODUZIONE
3
1.2. EFFETTI DEL CALORE SULLA COMPOSIZIONE DEL LATTE [1]
I primi tentativi a carattere scientifico sull’uso del calore per la conservazione
alimentare furono effettuati dal francese Appert [2], il quale dimostrò che taluni
alimenti posti in contenitori a chiusura ermetica e trattati termicamente si potevano
conservare per lungo tempo senza eccessive modificazioni delle proprietà igieniche,
organolettiche e nutritive.
Gli effetti del trattamento termico riguardano le proprietà chimico-fisiche e
organolettiche del latte. Il calore determina alterazioni di tali caratteristiche in
maniera proporzionale alla temperatura e al tempo di esposizione. Le modificazioni
variano in funzione del sistema di riscaldamento e delle modalità dell’intero ciclo di
trattamento [3].
1.2.1. PROPRIETÀ ORGANOLETTICHE
L’azione del calore sul latte ha inizio a 40-50 °C, facilitando l’eliminazione dei
gas disciolti e formazione di una pellicola. Il sapore di cotto, caratteristico del latte
termizzato, dipende dalla temperatura; con un trattamento fino a 60 °C il sapore di
cotto comincia a presentarsi, a 70 °C si rende più manifesto e con temperature di 80 °C
si forma quasi istantaneamente. Parallelamente a questo fenomeno si manifesta
l’odore di cotto dovuto a sostanze volatili come idrogeno solforato, ammoniaca,
composti fosforati e mercaptani, che si liberano per termolisi delle molecole
proteiche. L’adozione di tecniche quali la refrigerazione e l’areazione, permette
l’allontanamento di tali sostanze dal latte.
1-INTRODUZIONE
4
A temperature elevate, superiori ai 100 °C, si ottiene un’alterazione della
normale colorazione del latte dovuta a reazioni di imbrunimento non enzimatico
(reazione di Maillard) che coinvolgono gli zuccheri e le proteine del latte.
1.2.2. PROPRIETÀ CHIMICO - NUTRIZIONALI
L’effetto del trattamento termico sulle sostanze proteiche è costituito da una
parziale idrolisi, con relativo aumento di amminoacidi liberi. In particolare aumenta
la digeribilità della caseina, poiché la coagulazione avviene sotto forma di particelle
molto piccole. Le proteine del siero iniziano ad alterarsi a circa 65 °C, essendo più
sensibili all’azione del calore rispetto alle caseine.
La sostanza grassa, durante i normali trattamenti termici, non risente di
particolari modificazioni chimiche. La quantità di calcio solubile si riduce per effetto
del calore poiché viene influenzato lo stato di dispersione colloidale del fosfato di
calcio. Le vitamine contenute nel latte sono relativamente resistenti ai trattamenti
termici usuali, vengono ridotte in parte la vitamina C, le vitamine B1 e B2. Il lattosio
si ossida e caramellizza intorno ai 130 °C, subendo la reazione di Maillard.
1-INTRODUZIONE
5
1.3. IL LATTULOSIO
La possibilità di possedere degli indici di riferimento affidabili per determinare
l’entità del trattamento termico, è un obbiettivo molto ambito. Diversi sono stati i
tentativi negli ultimi vent’anni; sono stati proposti almeno tre diversi indicatori basati
sulla determinazione della BSA (una sieroproteina del latte), della furosina e del
lattulosio [4].
Il lattulosio è un disaccaride costituito da galattosio e fruttosio. Esso è totalmente
assente nel latte crudo, in quanto si forma dall’epimerizzazione del lattosio durante il
trattamento termico del latte alimentare [5]. Secondo la nomenclatura IUPAC, il
lattulosio è un 4-O-β-D-galattopiranosil-D-fruttosio (Figura1.1).
180 ms), ERED = -220 mV (tRED = 360 ms). La corrente risultante dall’ossidazione degli
analiti è stata campionata ed integrata negli ultimi 200 ms dell’applicazione del
potenziale di determinazione per minimizzare la corrente di carica. La centrifuga
utilizzata per la preparazione dei campioni è una A.L.C. 4525 (Apparecchiature per
Laboratori Chimici s.r.l.).
3-TECNICHE SPERIMENTALI
24
3. TECNICHE SPERIMENTALI
3.1. CROMATOGRAFIA
La cromatografia comprende tutta una serie di tecniche atte separare i soluti
presenti in una miscela, al fine di una loro identificazione, quantificazione ed
eventuale purificazione.
Le basi teoriche che governano i processi cromatografici, sono basate
sull’equilibrio che i soluti instaurano con le due fasi: la fase mobile e la fase
stazionaria. La fase mobile in cui viene disciolto l’analita può essere un gas, un
liquido o un fluido supercritico; essa viene fatta passare attraverso un supporto, che
può essere una colonna o una superficie solida, contenente la fase stazionaria. Questa
dovrà essere immiscibile con la fase mobile e dovrà essere scelta in modo che la
miscela dei soluti che viene iniettata si ripartisca tra le due fasi in funzione delle
caratteristiche di ogni singolo componente. I soluti che avranno maggiore affinità
con la fase stazionaria rimarranno più tempo nella colonna cromatografica di quelli
con minore affinità. In conseguenza di queste differenze i composti presenti nel
campione si separeranno in bande discrete e potranno quindi essere analizzati
quantitativamente e qualitativamente.
Le anzidette bande possono essere registrate per mezzo di opportuni rivelatori
che misurano un segnale in funzione del tempo. Questi diagrammi prendono il nome
di cromatogrammi ed i singoli segnali relativi ad ogni analita sono denominati
picchi.
3-TECNICHE SPERIMENTALI
25
3.2. PARAMETRI CROMATOGRAFICI
Per descrivere analiticamente le separazioni cromatografiche sono usualmente
utilizzati i seguenti parametri:
3.2.1. NUMERO DI PIATTI TEORICI
Il numero di piatti teorici è una misura quantitativa dell’efficienza di una
colonna; il parametro viene generalmente valutato sia mediante la comune
espressione
che con la relazione proposta da Foley e Dorsey [23]
nel caso di picchi asimmetrici, dove tr è il tempo di ritenzione (min.), w0.5 e w0.1
rappresentano rispettivamente le ampiezze al 50 e al 10% di altezza del picco
cromatografico, mentre As è il fattore di asimmetria, B/A; quando B≥Α, dove B ed
A sono rispettivamente le semi ampiezze destra e sinistra calcolate al 10% di altezza
del picco.
2
5.0
545,5
=
wtN r
25,1
1,472
1.01.0 +
=s
r
Awt
N
3-TECNICHE SPERIMENTALI
26
3.2.2. FATTORE DI CAPACITÀ
Il fattore di capacità è calcolato con la seguente equazione:
dove t0 = tempo morto della colonna
3.2.3. FATTORE DI SELETTIVITÀ
Il fattore di selettività è definito come:
dove t’r1 e t’r2 (t’r2 > t’r1) sono i cosiddetti tempi di ritenzione corretti per il tempo
morto (t’r=tr-t0) riferiti rispettivamente a due picchi consecutivi.
3.2.4. RISOLUZIONE
Per il calcolo della risoluzione è stata utilizzata la seguente formula:
Con wa e wb si indicano le larghezze dei picchi alla base, mentre con tr i tempi di
ritenzione.
0
0't
ttk r −
=
1
2
1
2
01
02
''
kk
tt
tttt
r
r
r
r ==−−
=α
( ) ( )[ ]ba
arbr
wwttRs
+−×= 2
3-TECNICHE SPERIMENTALI
27
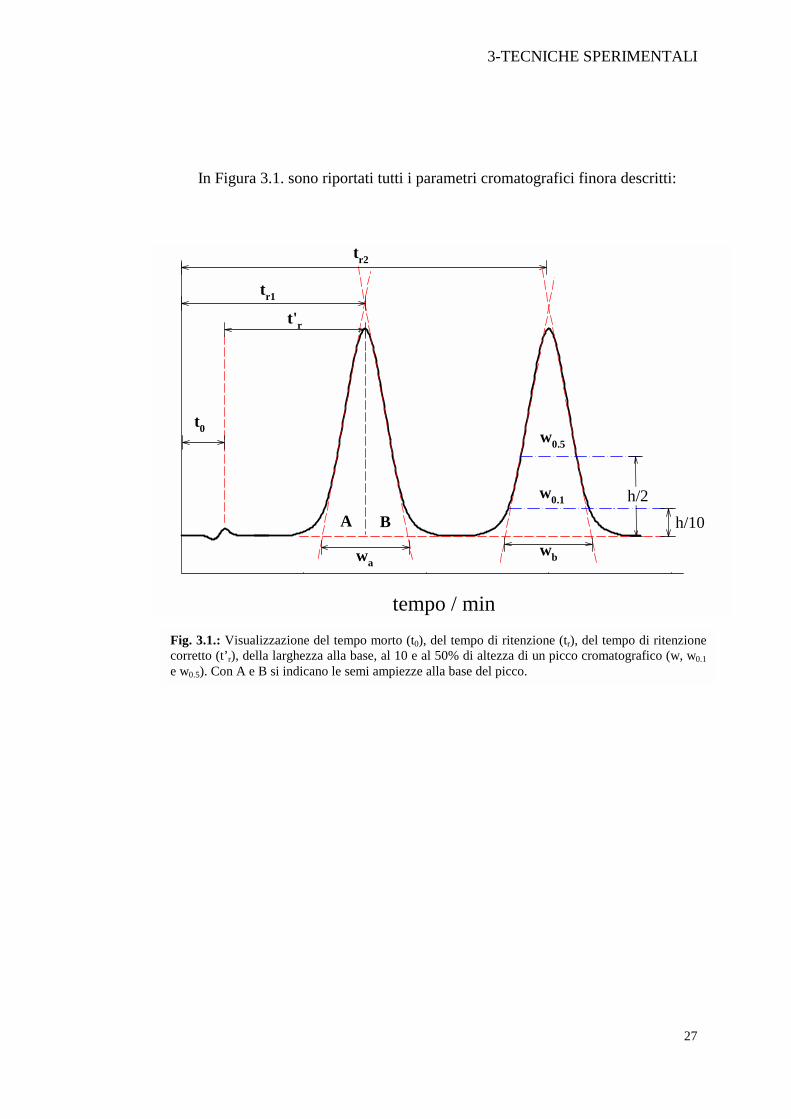
In Figura 3.1. sono riportati tutti i parametri cromatografici finora descritti:
tempo / min
t0
wawb
tr2
tr1
t'r
A Bh/2
h/10
w0.1
w0.5
Fig. 3.1.: Visualizzazione del tempo morto (t0), del tempo di ritenzione (tr), del tempo di ritenzionecorretto (t’r), della larghezza alla base, al 10 e al 50% di altezza di un picco cromatografico (w, w0.1e w0.5). Con A e B si indicano le semi ampiezze alla base del picco.
3-TECNICHE SPERIMENTALI
28
3.3. CROMATOGRAFIA LIQUIDA AD ALTE PRESTAZIONI
La cromatografia liquida ad alte prestazioni è una delle tecniche più diffuse
per la determinazione dei carboidrati negli alimenti. La sigla HPAEC-PAD, è
l’acronimo di High-Performance Anion-Exchange Chromatography coupled with
Pulsed Amperometric Detection, con il quale viene indicata la tecnica HPLC che
utilizza una fase stazionaria a scambio anionico con rivelazione amperometrica
pulsata. Le separazioni cromatografiche di carboidrati basate sullo scambio ionico
sono ottenute utilizzando fasi stazionarie a base polimerica funzionalizzate con
scambiatori anionici. La ritenzione selettiva di specie cariche in un sistema a scambio
ionico avviene a causa delle differenti interazioni che hanno luogo tra gli analiti ed il
gruppo carico con il quale è stato funzionalizzata la fase stazionaria.
I gruppi che caratterizzano la fase stazionaria possono essere distinti in:
♦ scambiatori cationici (gruppi carichi negativamente come es. quello
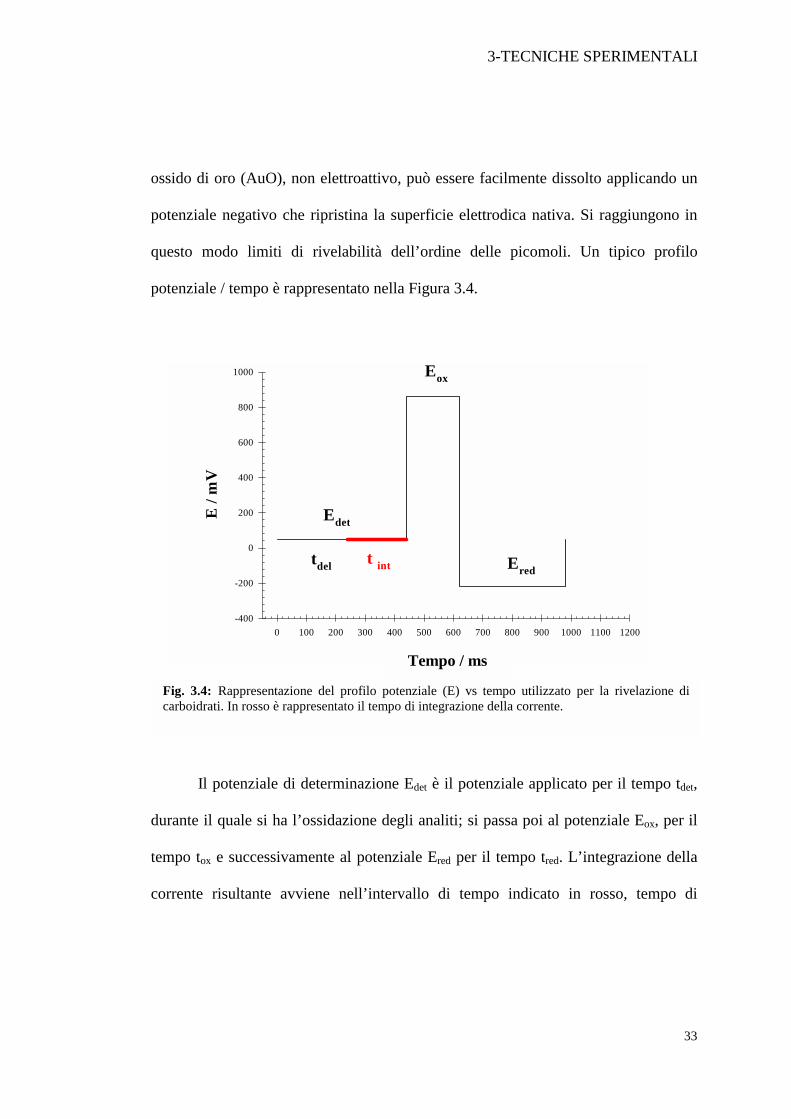
Fig. 3.4: Rappresentazione del profilo potenziale (E) vs tempo utilizzato per la rivelazione di carboidrati. In rosso è rappresentato il tempo di integrazione della corrente.
Tempo / ms
3-TECNICHE SPERIMENTALI
34
integrazione (tint). L’attesa del tempo tdel dopo il passaggio del potenziale da Ered a
Edet evita di registrare la corrente di carica dell’elettrodo.
I valori dei potenziali da applicare dipendono dalle condizioni sperimentali e
dagli analiti da rivelare. In genere la scelta è basata su prove in voltammetria ciclica [18].
Nel presente lavoro di tesi la forma d’onda, come verrà descritto in seguito, è
stata ottimizzata sui seguenti valori:
Tabella 3.3: Potenziali e tempi di applicazione ottimizzati per la rivelazione di carboidrati relativi al profilo riportato in fig. 3.4.
Potenziale mV Tempo / ms
Edet 50 440(totale) 200(integrazione)
Eox 860 180
Ered -220 360
4-RISULTATI E DISCUSSIONE
35
4. RISULTATI E DISCUSSIONE
Nella parte introduttiva sono state elencate le tecniche classiche utilizzate per
la determinazione del lattulosio, è stata sottolineata la difficoltà legata alla quantità di
lattosio presente nel latte in confronto alle quantità di lattulosio e sono state messe in
evidenza quali sono le limitazioni di tali tecniche nelle analisi di routine.
Lo scopo del lavoro di tesi è stato quello di mettere a punto una metodica
capace di separare e determinare il lattulosio nel latte mediante l’ottimizzazione di
una tecnica di cromatografia liquida ad alte prestazioni accoppiata ad un rivelatore
amperometrico pulsato impiegando un elettrodo d’oro.
4.1. FASE MOBILE: CONCENTRAZIONE DELL’NaOH
La scelta della fase mobile, cioè la concentrazione di ioni idrossido che deve
essere utilizzata per eseguire una separazione con cromatografia a scambio anionico,
rappresenta la prima fase nella messa a punto di un metodo. La concentrazione di
OH- deve essere tale da assicurare la separazione degli zuccheri presenti nella
miscela in esame, mentre la separazione può avvenire con eluizione isocratica o in
gradiente. Come vedremo nel corso della descrizione del metodo, abbiamo scelto di
lavorare in condizioni isocratiche perché la tecnica proposta mira ad ottenere la
separazione di miscele di carboidrati del latte mediante l’utilizzazione di basse
concentrazioni di ioni idrossido, evitando di rigenerare la colonna dopo ogni
separazione. Con una separazione in gradiente, oltre agli inconvenienti noti
4-RISULTATI E DISCUSSIONE
36
riguardanti la deriva della linea di base, avremmo dovuto rigenerare e poi
condizionare la colonna dopo ogni analisi, il che avrebbe inficiato lo scopo del
presente lavoro.
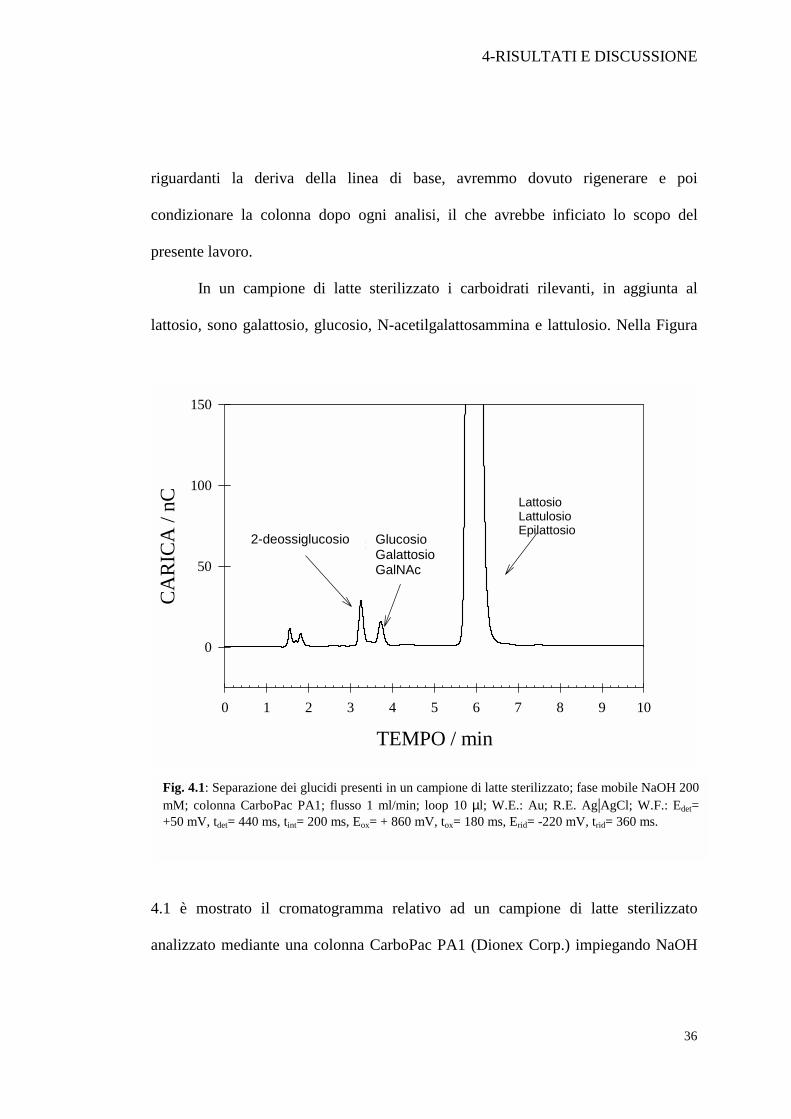
In un campione di latte sterilizzato i carboidrati rilevanti, in aggiunta al
lattosio, sono galattosio, glucosio, N-acetilgalattosammina e lattulosio. Nella Figura
4.1 è mostrato il cromatogramma relativo ad un campione di latte sterilizzato
analizzato mediante una colonna CarboPac PA1 (Dionex Corp.) impiegando NaOH
TEMPO / min0 1 2 3 4 5 6 7 8 9 10
CA
RIC
A /
nC
0
50
100
150
2-Deossiglucosio D-GlucosioD-GalattosioMannosio
LattosioLattulosioEpilattosio
Fig. 4.1: Separazione dei glucidi presenti in un campione di latte sterilizzato; fase mobile NaOH 200 mM; colonna CarboPac PA1; flusso 1 ml/min; loop 10 µl; W.E.: Au; R.E. Ag|AgCl; W.F.: Edet= +50 mV, tdet= 440 ms, tint= 200 ms, Eox= + 860 mV, tox= 180 ms, Erid= -220 mV, trid= 360 ms.
Glucosio Galattosio GalNAc
2-deossiglucosio
4-RISULTATI E DISCUSSIONE
37
200 mM come fase mobile e 2-deossiglucosio come standard interno. Come si può
vedere, anche se il tempo totale dell’analisi è contenuto entro i 7 minuti, il numero di
picchi cromatografici è inferiore al numero di componenti presenti nel campione di
latte, probabilmente a causa di una loro sovrapposizione. Il problema è da ricondursi
all’elevata concentrazione di ioni OH¯ che non assicura un’adeguata selettività della
separazione cromatografica.
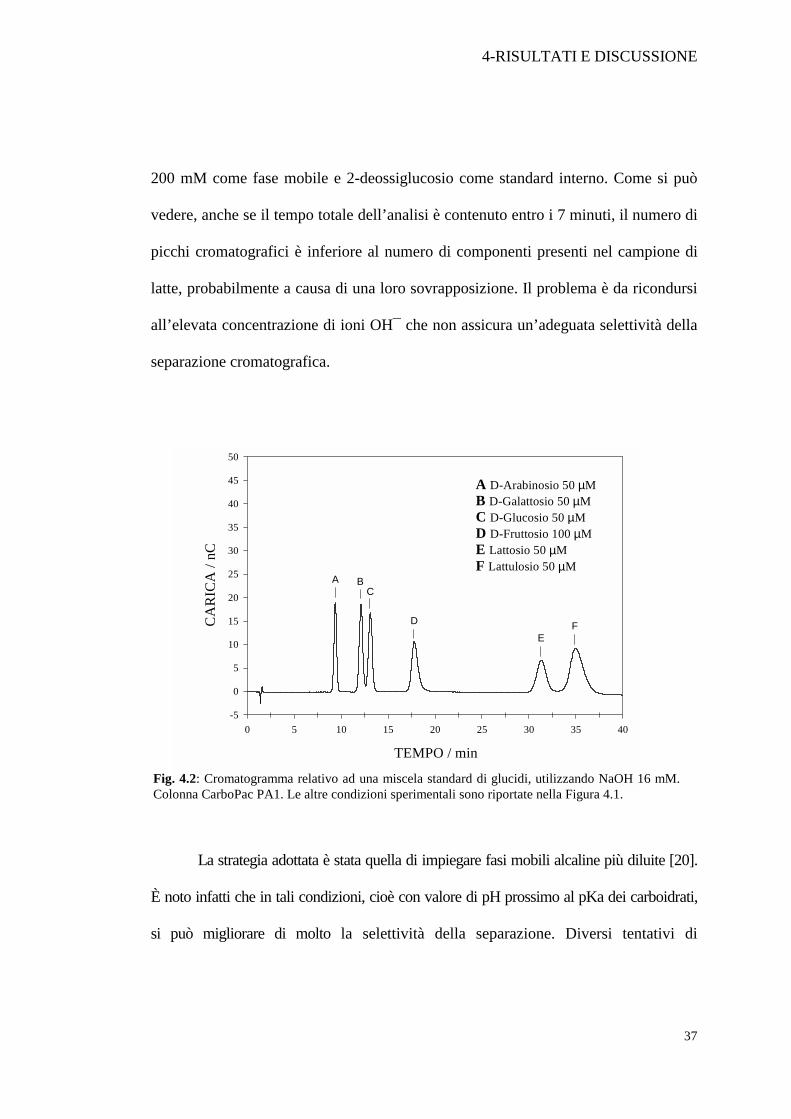
La strategia adottata è stata quella di impiegare fasi mobili alcaline più diluite [20].
È noto infatti che in tali condizioni, cioè con valore di pH prossimo al pKa dei carboidrati,
si può migliorare di molto la selettività della separazione. Diversi tentativi di
Fig. 4.2: Cromatogramma relativo ad una miscela standard di glucidi, utilizzando NaOH 16 mM. Colonna CarboPac PA1. Le altre condizioni sperimentali sono riportate nella Figura 4.1.
4-RISULTATI E DISCUSSIONE
38
separazione cromatografica sono stati eseguiti, adoperando una soluzione standard
contenente arabinosio, galattosio, glucosio, fruttosio, lattosio e lattulosio. L’indicazione
emersa è che solo l’impiego di soluzioni diluite di NaOH può consentire una buona
risoluzione. In Figura 4.2 è mostrato il cromatogramma ottenuto eluendo con una
soluzione di NaOH pari a 16 mM. Si può notare che la separazione è decisamente
migliorata per quasi tutti gli analiti ad eccezione del galattosio e glucosio che
presentano ancora una lieve sovrapposizione. Da notare anche che il miglioramento è
stato ottenuto a scapito di un notevole aumento nei tempi di ritenzione. Il lattulosio,
ad esempio, individuabile nell’ultimo picco della serie (picco F), appare ad un tempo
di ritenzione di circa 35 minuti. Nel tentativo di migliorare la risoluzione della
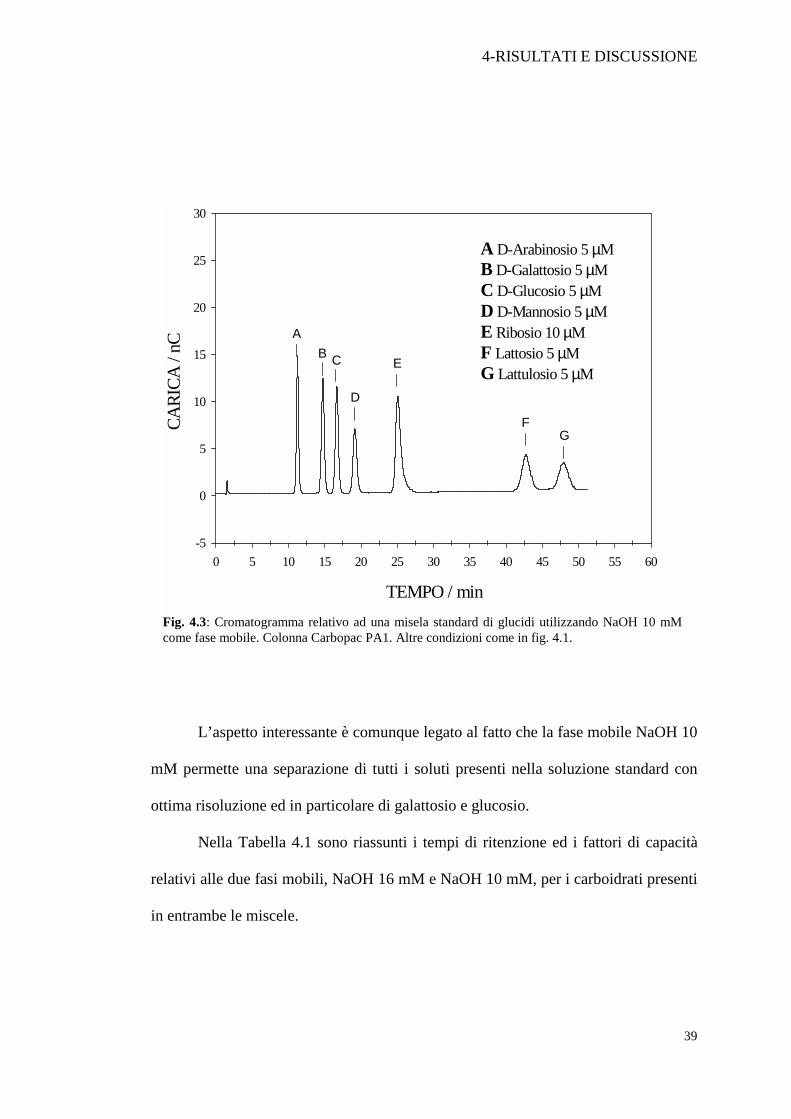
separazione, la concentrazione di NaOH è stata portata fino a 10 mM (Figura 4.3).
Questo ha comportato un ulteriore aumento dei tempi di ritenzione.
4-RISULTATI E DISCUSSIONE
39
L’aspetto interessante è comunque legato al fatto che la fase mobile NaOH 10
mM permette una separazione di tutti i soluti presenti nella soluzione standard con
ottima risoluzione ed in particolare di galattosio e glucosio.
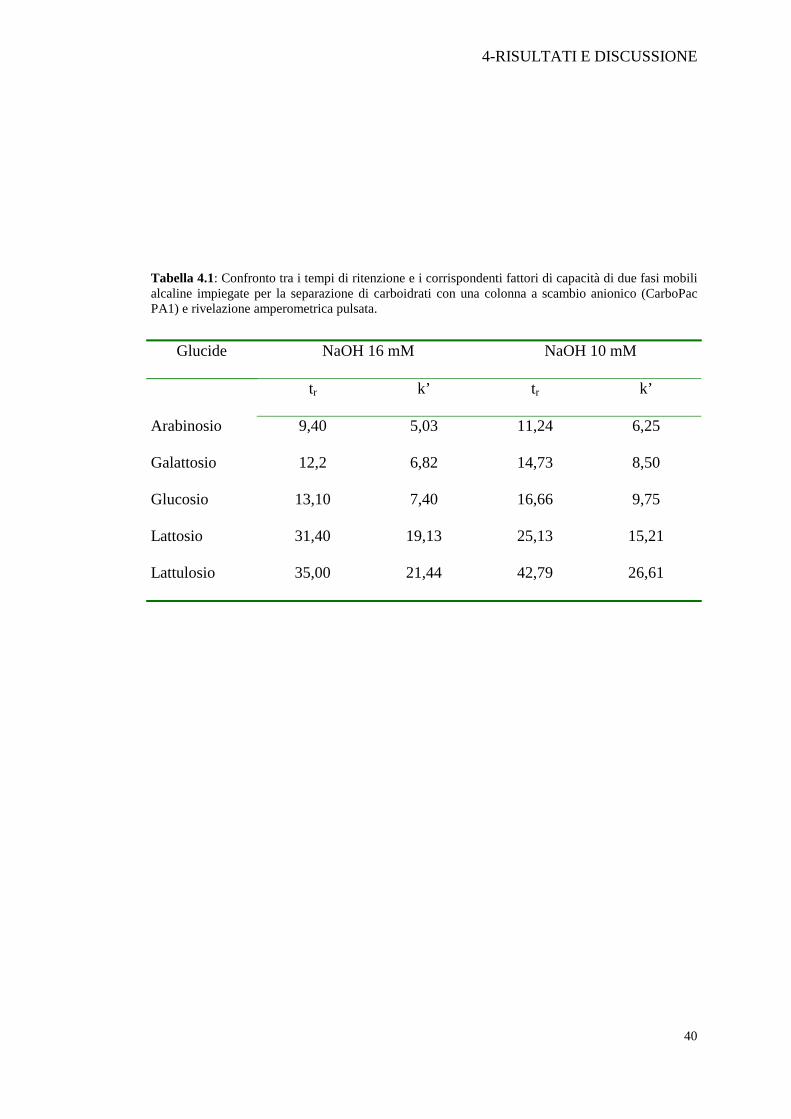
Nella Tabella 4.1 sono riassunti i tempi di ritenzione ed i fattori di capacità
relativi alle due fasi mobili, NaOH 16 mM e NaOH 10 mM, per i carboidrati presenti
Fig. 4.3: Cromatogramma relativo ad una misela standard di glucidi utilizzando NaOH 10 mMcome fase mobile. Colonna Carbopac PA1. Altre condizioni come in fig. 4.1.
4-RISULTATI E DISCUSSIONE
40
Tabella 4.1: Confronto tra i tempi di ritenzione e i corrispondenti fattori di capacità di due fasi mobili alcaline impiegate per la separazione di carboidrati con una colonna a scambio anionico (CarboPac PA1) e rivelazione amperometrica pulsata.
Glucide NaOH 16 mM NaOH 10 mM
tr k’ tr k’
Arabinosio 9,40 5,03 11,24 6,25
Galattosio 12,2 6,82 14,73 8,50
Glucosio 13,10 7,40 16,66 9,75
Lattosio 31,40 19,13 25,13 15,21
Lattulosio 35,00 21,44 42,79 26,61
4-RISULTATI E DISCUSSIONE
41
4.2. STABILITÀ DEI TEMPI DI RITENZIONE: EFFETTO DEL
CARBONATO
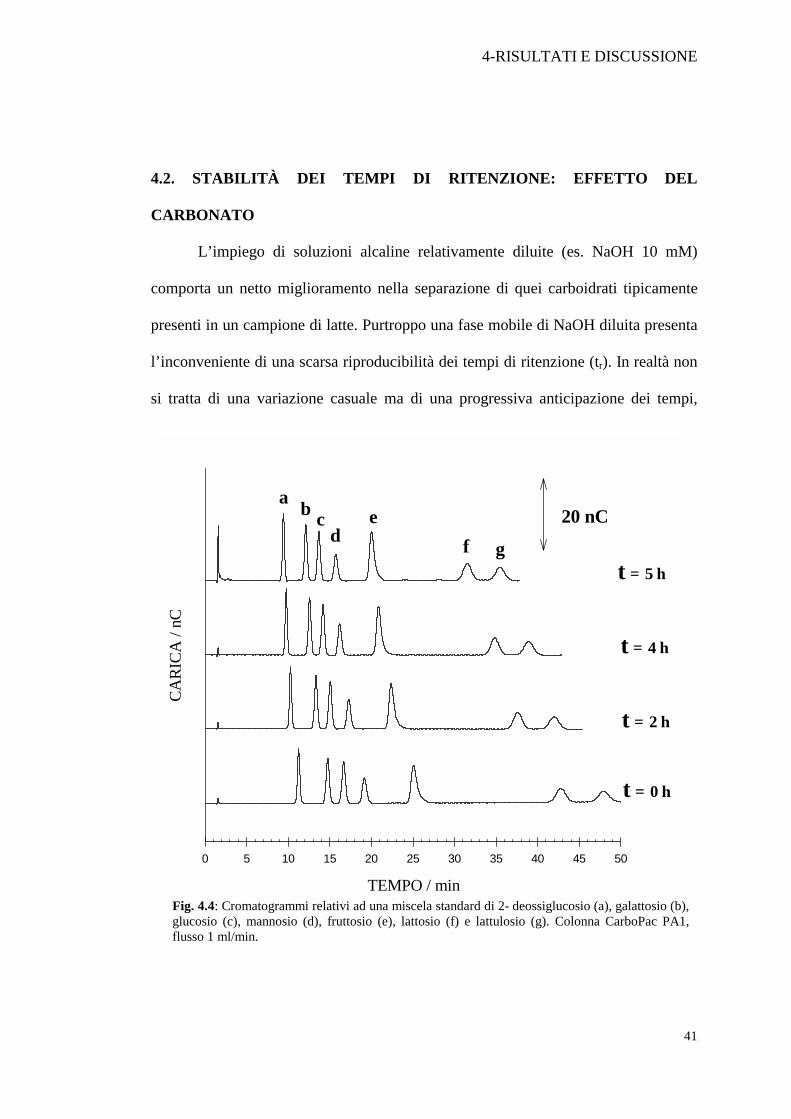
L’impiego di soluzioni alcaline relativamente diluite (es. NaOH 10 mM)
comporta un netto miglioramento nella separazione di quei carboidrati tipicamente
presenti in un campione di latte. Purtroppo una fase mobile di NaOH diluita presenta
l’inconveniente di una scarsa riproducibilità dei tempi di ritenzione (tr). In realtà non
si tratta di una variazione casuale ma di una progressiva anticipazione dei tempi,
TEMPO / min
0 5 10 15 20 25 30 35 40 45 50
CARI
CA /
nC
20 nC
t = 5 h
t = 4 h
t = 2 h
t = 0 h
a b cd
ef g
Fig. 4.4: Cromatogrammi relativi ad una miscela standard di 2- deossiglucosio (a), galattosio (b), glucosio (c), mannosio (d), fruttosio (e), lattosio (f) e lattulosio (g). Colonna CarboPac PA1, flusso 1 ml/min.
4-RISULTATI E DISCUSSIONE
42
come si può riscontrare in seguito a successive iniezioni di una soluzione standard. In
Figura 4.4 sono riportati una serie di cromatogrammi acquisiti a tempi diversi
impiegando come fase eluente una soluzione di NaOH 10 mM. Col passare del tempo la
colonna dimostra una sempre più bassa capacità di scambio, cioè una minore interazione con
i soluti. Questo fenomeno sembra essere causato da una progressiva occupazione dei siti di
scambio della colonna da parte degli ioni carbonato, inevitabilmente presenti nell’eluente
alcalino, ed originati dalla CO2 atmosferica disciolta. Dopo solo 5 ore di analisi, i tempi di
ritenzione di tutti i soluti si sono marcatamente ridotti; il lattulosio (picco g) ad esempio
presenta un tempo di ritenzione che risulta anticipato di circa 12 minuti. In queste condizioni
risulta abbastanza problematico eseguire un’analisi quali e quantitativa dei singoli soluti.
È opportuno sottolineare che i cromatogrammi in Figura 4.4 sono stati ottenuti
impiegando un eluente preparato in condizioni tali da limitare al massimo la dissoluzione di
anidride carbonica nella soluzione. In primo luogo è stato effettuato un prolungato
degasaggio dell’acqua con elio; inoltre la bottiglia contenente la fase mobile è stata sempre
tenuta pressurizzata con lo stesso gas inerte. In queste condizioni di lavoro, l’impiego di
soluzioni eluenti alcaline diluite non è del tutto precluso. In realtà, così come riportato dalla
Dionex [25], la metodica prevede tra una corsa e l’altra, la rigenerazione della colonna con
NaOH concentrata (0,2 ÷ 0,5 M) per circa 15 minuti e successivo condizionamento per un
periodo di tempo variabile dai 15 ai 30 minuti con la fase mobile di lavoro. Questa procedura
comporta ovviamente una notevole perdita di tempo nelle analisi di routine e una bassa
riproducibilità dei tempi di ritenzione tra due iniezioni intercalate dalla rigenerazione.
4-RISULTATI E DISCUSSIONE
43
4.3. STRATEGIE PER MIGLIORARE LA RIPRODUCIBILITÀ DEI TEMPI
DI RITENZIONE
Nel nostro laboratorio è stata di recente messa a punto una procedura
innovativa finalizzata a rimuovere in maniera semplice ed efficace gli ioni carbonato
dagli eluenti usati in cromatografia con colonne a scambio anionico (HPAEC). Tale
metodica consiste nell’aggiungere sali di cationi di metalli alcalino-terrosi, come
Ba2+, Sr2+, o Ca2+, alla fase mobile, inducendo così la precipitazione di carbonati
insolubili. Nella Tabella 4.2 sono riassunte le prove effettuate con diversi cationi
bivalenti e con concentrazioni comprese tra 0,5 e 2 mM.
Tabella 4.2: Riepilogo delle prove effettuate con i diversi cationi aggiunti in fase mobile alcalina.
Concentrazione di NaOH Composto aggiunto
Ba(OAc)2, 1 mM
Ba(OH)2, 1 mM 16 mM Ca(OAc)2, 0,5 mM
Ba(OAc)2, 1 e 2 mM
Sr(OAc)2, 1 e 2 mM 10 mM Zn(OAc)2, 1 mM
In primo luogo abbiamo rivolto la nostra attenzione alla scelta del catione in
grado di assicurare la rimozione più efficace del carbonato, inevitabilmente presente
nella fase mobile alcalina. In Tabella 4.3 sono riportate le solubilità in acqua degli
4-RISULTATI E DISCUSSIONE
44
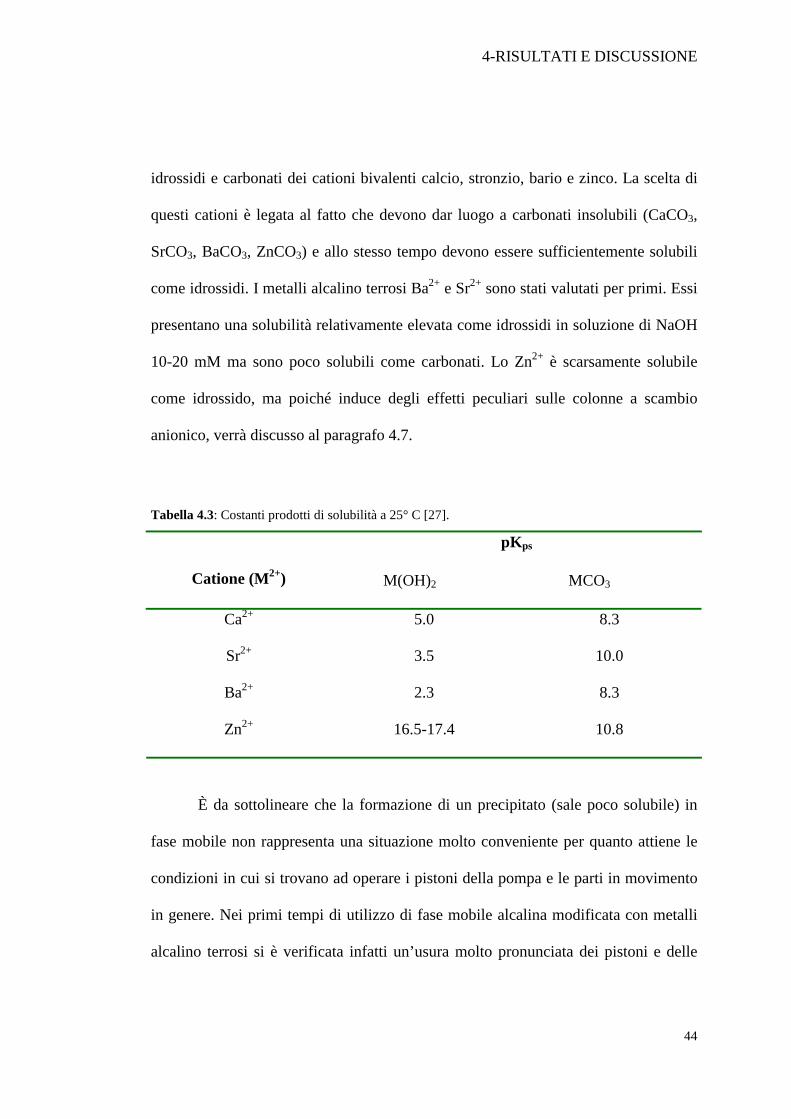
idrossidi e carbonati dei cationi bivalenti calcio, stronzio, bario e zinco. La scelta di
questi cationi è legata al fatto che devono dar luogo a carbonati insolubili (CaCO3,
SrCO3, BaCO3, ZnCO3) e allo stesso tempo devono essere sufficientemente solubili
come idrossidi. I metalli alcalino terrosi Ba2+ e Sr2+ sono stati valutati per primi. Essi
presentano una solubilità relativamente elevata come idrossidi in soluzione di NaOH
10-20 mM ma sono poco solubili come carbonati. Lo Zn2+ è scarsamente solubile
come idrossido, ma poiché induce degli effetti peculiari sulle colonne a scambio
anionico, verrà discusso al paragrafo 4.7.
Tabella 4.3: Costanti prodotti di solubilità a 25° C [27].
pKps
Catione (M2+) M(OH)2 MCO3
Ca2+ 5.0 8.3
Sr2+ 3.5 10.0
Ba2+ 2.3 8.3
Zn2+ 16.5-17.4 10.8
È da sottolineare che la formazione di un precipitato (sale poco solubile) in
fase mobile non rappresenta una situazione molto conveniente per quanto attiene le
condizioni in cui si trovano ad operare i pistoni della pompa e le parti in movimento
in genere. Nei primi tempi di utilizzo di fase mobile alcalina modificata con metalli
alcalino terrosi si è verificata infatti un’usura molto pronunciata dei pistoni e delle
4-RISULTATI E DISCUSSIONE
45
guarnizioni. È stato però rilevato con grande soddisfazione che se l’aggiunta di bario,
stronzio o calcio acetato alla fase mobile alcalina veniva effettuata almeno un giorno
prima del suo impiego, l’eventuale formazione di precipitato (CaCO3, SrCO3,
BaCO3) ha la possibilità di depositarsi sul fondo della riserva di fase mobile,
evitando danni alle parti meccaniche in movimento della pompa e dell’iniettore.
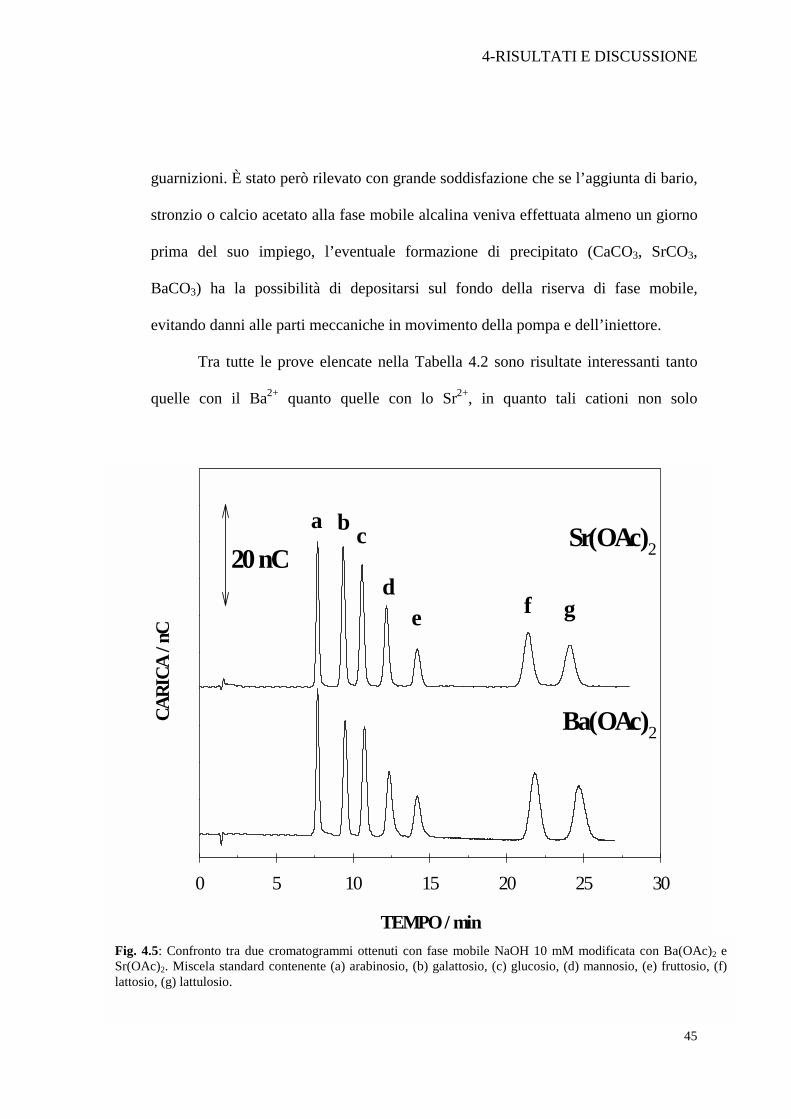
Tra tutte le prove elencate nella Tabella 4.2 sono risultate interessanti tanto
quelle con il Ba2+ quanto quelle con lo Sr2+, in quanto tali cationi non solo
TEMPO / min
0 5 10 15 20 25 30
CARI
CA /
nC
Sr(OAc)2
Ba(OAc)2
20 nCa b c
de f g
Fig. 4.5: Confronto tra due cromatogrammi ottenuti con fase mobile NaOH 10 mM modificata con Ba(OAc)2 e Sr(OAc)2. Miscela standard contenente (a) arabinosio, (b) galattosio, (c) glucosio, (d) mannosio, (e) fruttosio, (f) lattosio, (g) lattulosio.
4-RISULTATI E DISCUSSIONE
46
garantiscono un’ottima riproducibilità dei tempi di ritenzione, ma inducono anche un
aumento del responso amperometrico, come verrà discusso nella parte 4.4. Nella
Figura 4.5 sono posti a confronto due cromatogrammi ottenuti con una fase mobile
NaOH 10 mM modificata con bario acetato o stronzio acetato, relativi ad una miscela
standard contenente arabinosio (a), galattosio (b), glucosio (c), mannosio (d),
fruttosio (e), lattosio (f) e lattulosio (g). Come si può notare sia i tempi di ritenzione
che l’intensità dei singoli analiti sono paragonabili.
La maggior parte degli esperimenti riportati in questo lavoro sono stati
eseguiti con aggiunta alla fase mobile alcalina di Ba(OAc)2, perché il bario porta alla
formazione di BaCO3 meno solubile di SrCO3 (Tabella 4.3). L’aggiunta del bario
sotto forma di Ba(OH)2 non si è dimostrata molto efficace. Infatti l’impiego di bario
idrossido comporta la formazione di una soluzione che rimane torbida anche per
diverse ore dopo l’aggiunta, probabilmente perché l’equilibrio eterogeneo di
dissoluzione del Ba(OH)2 è lento (vedi schema seguente):
L’aggiunta in fase mobile alcalina di Ba(OAc)2 alla concentrazione 1-2 mM
è sembrata la scelta più valida, in considerazione anche del fatto che
l’acetato è un forte controione e gioca un ruolo importante nella riduzione
dei tempi di ritenzione.
Ba(OH)2 s Ba(OH)2 aq Ba + 2OH2+ -
+CO3
2-BaCO3 aqBaCO3
4-RISULTATI E DISCUSSIONE
47
La migliore selettività per la separazione dei carboidrati testati è stata
ottenuta attraverso una fase mobile composta da NaOH 10 mM + Ba(OAc)2;
pertanto, essa è stata utilizzata per la determinazione del lattulosio nei
campioni di latte analizzati.
Per dimostrare l’efficacia della scelta sono state fatte numerose prove
di riproducibilità. L’unica precauzione adottata, oltre alla filtrazione e
degasaggio (per circa 30 minuti con He) dell’acqua utilizzata per la
preparazione della fase eluente, è consistita nel preparare la fase mobile la
sera precedente il suo impiego. La riserva di fase mobile veniva inoltre
protetta dalla CO2 dell’aria tenendola saturata con He per tutta la durata della
prova. La colonna utilizzata è stata rigenerata ogni mattina per 20 minuti con
NaOH 200 mM e poi condizionata con la fase mobile per 40 min. Nella
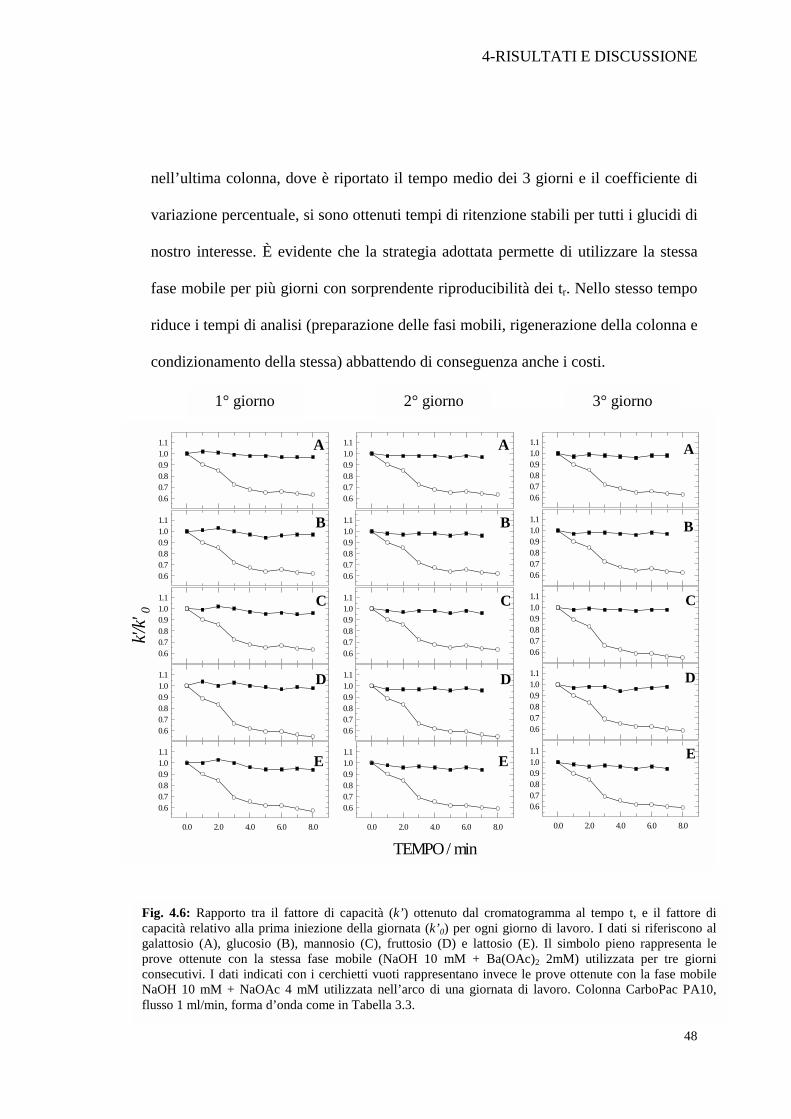
Figura 4.6 sono riportati i rapporti tra il fattore di capacità (k’) di galattosio
(A), glucosio (B), mannosio (C), fruttosio (D) e lattosio (E) ottenuti dal
cromatogramma registrato al tempo t, e i fattori di capacità relativi alla prima
iniezione della giornata (k’0). La stessa fase mobile costituita da NaOH 10
mM + Ba(OAc)2 2 mM è stata utilizzata per tre giorni consecutivi (cerchi
pieni). I simboli a cerchio vuoto fanno riferimento ai risultati ottenuti
utilizzando NaOH 10 mM + NaOAc 4 mM come eluente nell’arco di un solo
giorno di lavoro. I dati numerici delle prove precedenti e per altri glucidi
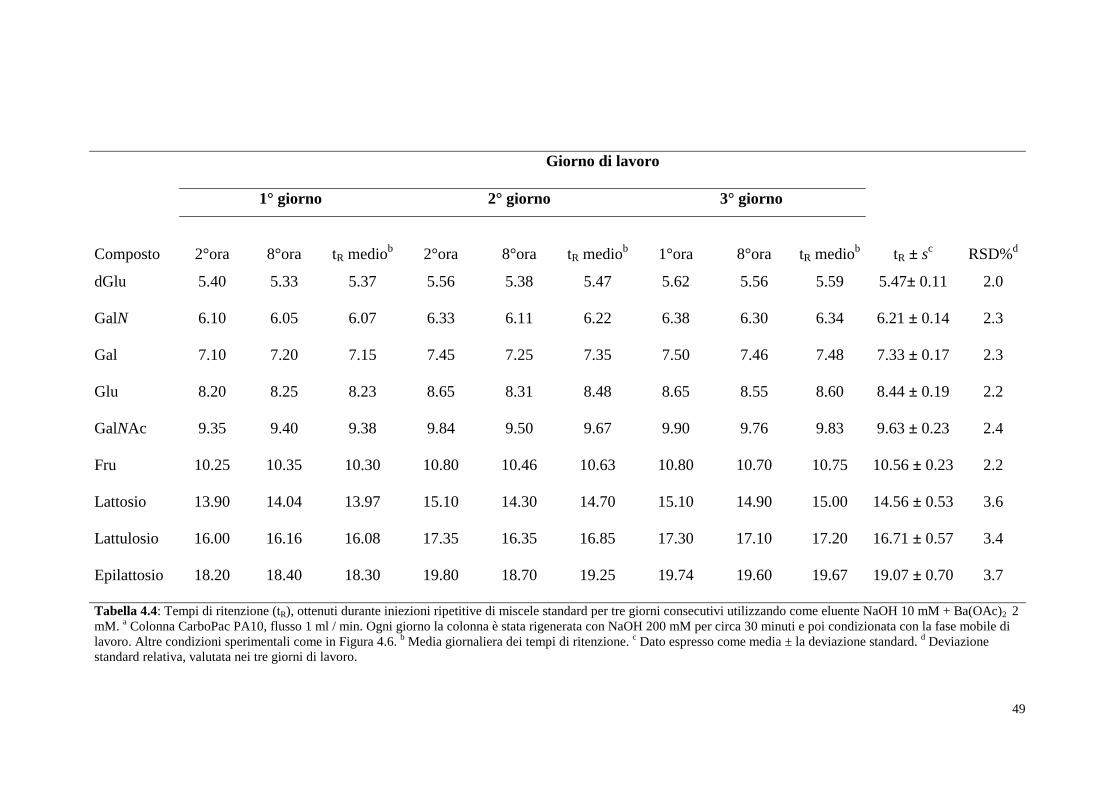
sono riportati nella Tabella 4.4 dove sono mostrati i tempi di ritenzione ad
inizio e fine giornata ed il tempo medio per ogni giorno. Come si può notare
4-RISULTATI E DISCUSSIONE
48
nell’ultima colonna, dove è riportato il tempo medio dei 3 giorni e il coefficiente di
variazione percentuale, si sono ottenuti tempi di ritenzione stabili per tutti i glucidi di
nostro interesse. È evidente che la strategia adottata permette di utilizzare la stessa
fase mobile per più giorni con sorprendente riproducibilità dei tr. Nello stesso tempo
riduce i tempi di analisi (preparazione delle fasi mobili, rigenerazione della colonna e
condizionamento della stessa) abbattendo di conseguenza anche i costi.
0.60.70.80.91.01.1 A
0.60.70.80.91.01.1
k'/k'
0
0.60.70.80.91.01.1
0.60.70.80.91.01.1
0.0 2.0 4.0 6.0 8.0
0.60.70.80.91.01.1
B
C
D
E
0.60.70.80.91.01.1 A
0.60.70.80.91.01.1
k'/k'
0
0.60.70.80.91.01.1
0.60.70.80.91.01.1
TEMPO / min0.0 2.0 4.0 6.0 8.0
0.60.70.80.91.01.1
B
C
D
E
0.60.70.80.91.01.1 A
0.60.70.80.91.01.1
k'/k'
0
0.60.70.80.91.01.1
0.60.70.80.91.01.1
TEMPO / min0.0 2.0 4.0 6.0 8.0
0.60.70.80.91.01.1
B
C
D
E
Fig. 4.6: Rapporto tra il fattore di capacità (k’) ottenuto dal cromatogramma al tempo t, e il fattore di capacità relativo alla prima iniezione della giornata (k’0) per ogni giorno di lavoro. I dati si riferiscono al galattosio (A), glucosio (B), mannosio (C), fruttosio (D) e lattosio (E). Il simbolo pieno rappresenta le prove ottenute con la stessa fase mobile (NaOH 10 mM + Ba(OAc)2 2mM) utilizzata per tre giorni consecutivi. I dati indicati con i cerchietti vuoti rappresentano invece le prove ottenute con la fase mobile NaOH 10 mM + NaOAc 4 mM utilizzata nell’arco di una giornata di lavoro. Colonna CarboPac PA10, flusso 1 ml/min, forma d’onda come in Tabella 3.3.
Tabella 4.4: Tempi di ritenzione (tR), ottenuti durante iniezioni ripetitive di miscele standard per tre giorni consecutivi utilizzando come eluente NaOH 10 mM + Ba(OAc)2 2 mM. a Colonna CarboPac PA10, flusso 1 ml / min. Ogni giorno la colonna è stata rigenerata con NaOH 200 mM per circa 30 minuti e poi condizionata con la fase mobile di lavoro. Altre condizioni sperimentali come in Figura 4.6. b Media giornaliera dei tempi di ritenzione. c Dato espresso come media ± la deviazione standard. d Deviazione standard relativa, valutata nei tre giorni di lavoro.
4-RISULTATI E DISCUSSIONE
50
4.4 EFFETTO DEI CATIONI SULLA RIVELAZIONE AMPEROMETRICA
PULSATA
L’utilizzazione di fasi mobili alcaline si adatta molto bene non solo per la
separazione dei glucidi in cromatografia a scambio anionico (HPAEC) ma anche per
la loro rilevazione mediante amperometria pulsata [26]. Questa tecnica prevede
l’aggiunta post-colonna di una base concentrata per migliorare la stabilità della linea
di base e la sensibilità del rivelatore, specialmente quando si utilizzano fasi mobili
neutre o con basso contenuto di OH- (come nel nostro caso) o comunque quando la
concentrazione di ioni idrossido è ≤ 20 mM. Recentemente è stato studiato l’effetto
di alcuni cationi divalenti non elettroattivi, come Sr(II), Ba(II) e Ca(II),
sull’ossidazione elettrochimica di alditoli e carboidrati su un elettrodo di oro in
amperometria pulsata [28] impiegando soluzioni di NaOH relativamente più
concentrate di quelle utilizzate nel presente lavoro di tesi. In particolare gli ioni
stronzio e bario, hanno evidenziato un miglioramento della sensibilità nella
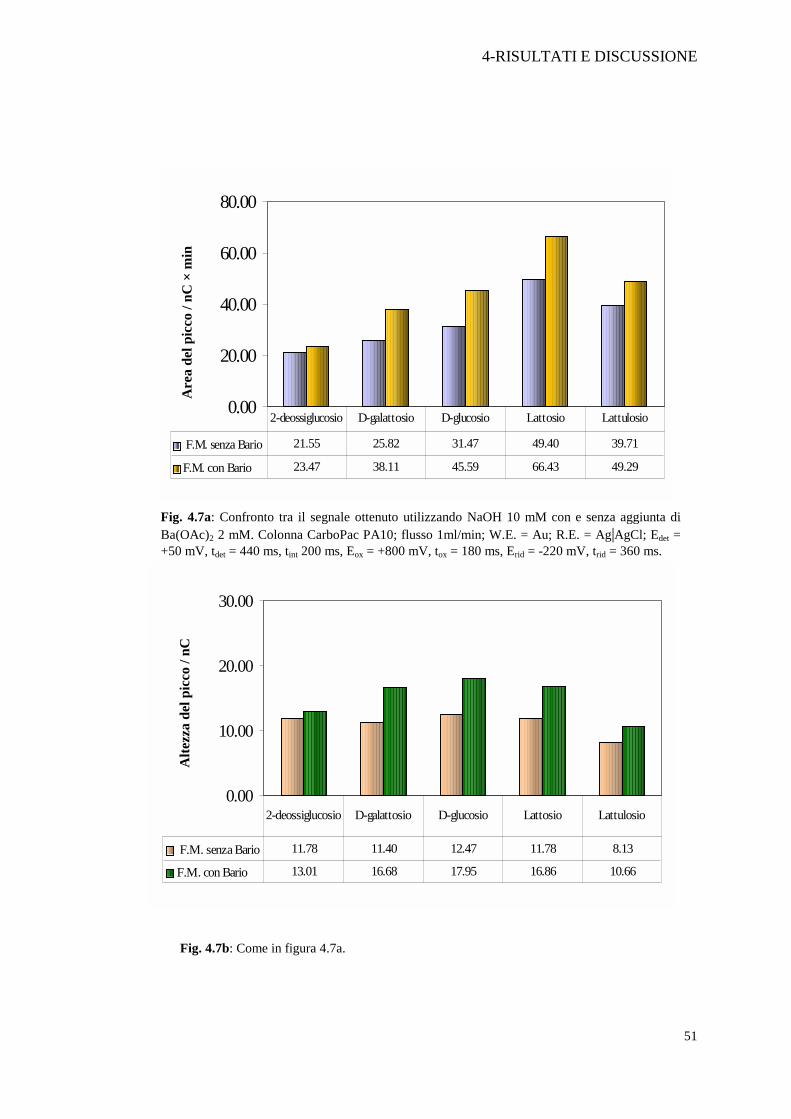
rivelazione. Questo effetto è stato confermato nelle nostre condizioni sperimentali
(Figura 4.7a e 4.7b). È palese infatti che sia quando si valuta la risposta degli analiti
in base all’area, sia in base all’altezza del picco la risposta in presenza del bario
aumenta in maniera evidente. Inoltre, come verrà mostrato in seguito, non è stato
necessario far ricorso ad aggiunte post-colonna poiché, pur lavorando con
concentrazioni 10 mM di NaOH, la sensibilità del metodo e la stabilità della linea di
base sono state assolutamente accettabili e in grado di garantire una buona
Fig. 4.7a: Confronto tra il segnale ottenuto utilizzando NaOH 10 mM con e senza aggiunta di Ba(OAc)2 2 mM. Colonna CarboPac PA10; flusso 1ml/min; W.E. = Au; R.E. = Ag|AgCl; Edet = +50 mV, tdet = 440 ms, tint 200 ms, Eox = +800 mV, tox = 180 ms, Erid = -220 mV, trid = 360 ms.
Fig. 4.7b: Come in figura 4.7a.
4-RISULTATI E DISCUSSIONE
52
4.5. ANALISI DI UN CAMPIONE REALE: TRATTAMENTO DEL
CAMPIONE DI LATTE
In considerazione degli ottimi risultati avuti impiegando come fase mobile
una soluzione diluita di NaOH addizionata opportunamente con bario acetato, sia in
termini di selettività della separazione che di durata della corsa cromatografica,
sensibilità e soprattutto riproducibilità dei tr, si è provato ad analizzare un campione
di latte nelle stesse condizioni sperimentali di Figura 4.6.
L’analisi di una matrice complessa come il latte necessita dapprima di un
pretrattamento del campione in modo tale da allontanare grassi e proteine. Sono stati
applicati due metodi di pretrattamento del campione, uno che prevede l’impiego di
etanolo come suggerito da Martinez e collaboratori e il secondo che fa ricorso all’uso
dei reattivi di Carrez. Di seguito sono descritti nel dettaglio i risultati dei due
procedimenti.
A: Etanolo; il trattamento con etanolo di un campione di latte oltre ad
allontanare proteine e grassi è riportato favorire una precipitazione selettiva del
lattosio [29, 30].
B: reattivi di Carrez; il trattamento del campione con i reagenti di Carrez I e
II determina una efficace deproteinizzazione ed allontanamento dei grassi dal
campione [31, 32, 33].
4-RISULTATI E DISCUSSIONE
53
TEMPO / min
0 5 10 15 20
A
B
1 2
3
4
5
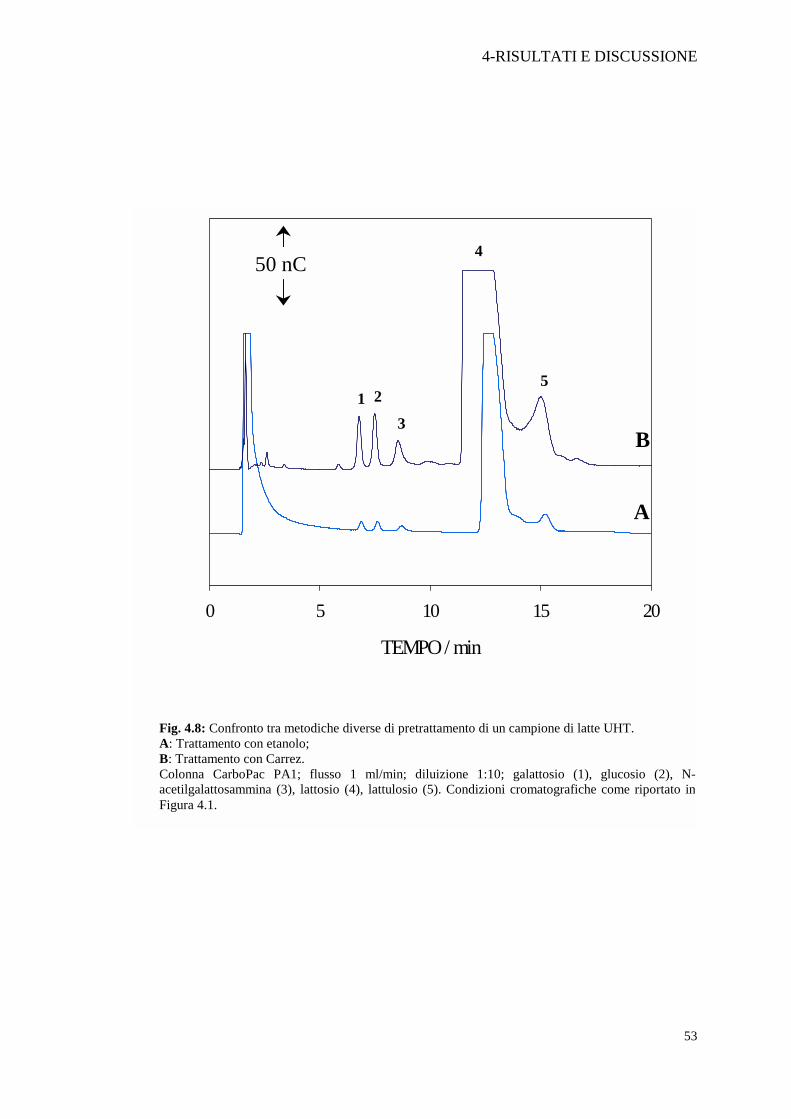
Fig. 4.8: Confronto tra metodiche diverse di pretrattamento di un campione di latte UHT. A: Trattamento con etanolo; B: Trattamento con Carrez. Colonna CarboPac PA1; flusso 1 ml/min; diluizione 1:10; galattosio (1), glucosio (2), N-acetilgalattosammina (3), lattosio (4), lattulosio (5). Condizioni cromatografiche come riportato in Figura 4.1.
50 nC
4-RISULTATI E DISCUSSIONE
54
Nella Figura 4.8 è mostrato il confronto tra i due diversi pretrattamenti valutati su
uno stesso campione di latte UHT di qualità Discount. La diluizione finale del
campione è stata di 1:10. Il trattamento con i reattivi di Carrez dimostra una migliore
“efficacia di estrazione” degli zuccheri del latte rispetto al trattamento con etanolo.
Quest’ultimo provoca un consistente calo del segnale, come si può notare dal
confronto dei cromatogrammi A e B in Figura 4.8, e non consente di ottenere un
cromatogramma “pulito” anche nei primi minuti d’analisi. A tal proposito si è anche
provato ad allontanare l’etanolo con He e ricostituendo la soluzione con acqua, ma la
prova non ha rivelato miglioramenti sostanziali.
È stato peraltro verificato se il trattamento con Carrez riduce o meno la
percentuale dei carboidrati presenti nel campione. La verifica è stata eseguita mediante
l’aggiunta di un composto standard ad un’aliquota di latte, utilizzando un carboidrato
notoriamente assente nella matrice lattea. Sono stati provati il 2-deossiglucosio, il 2-
deossiribosio, e il 3-ossimetilglucosio. Il 2-deossiglucosio (Figura 4.9) è sembrata la
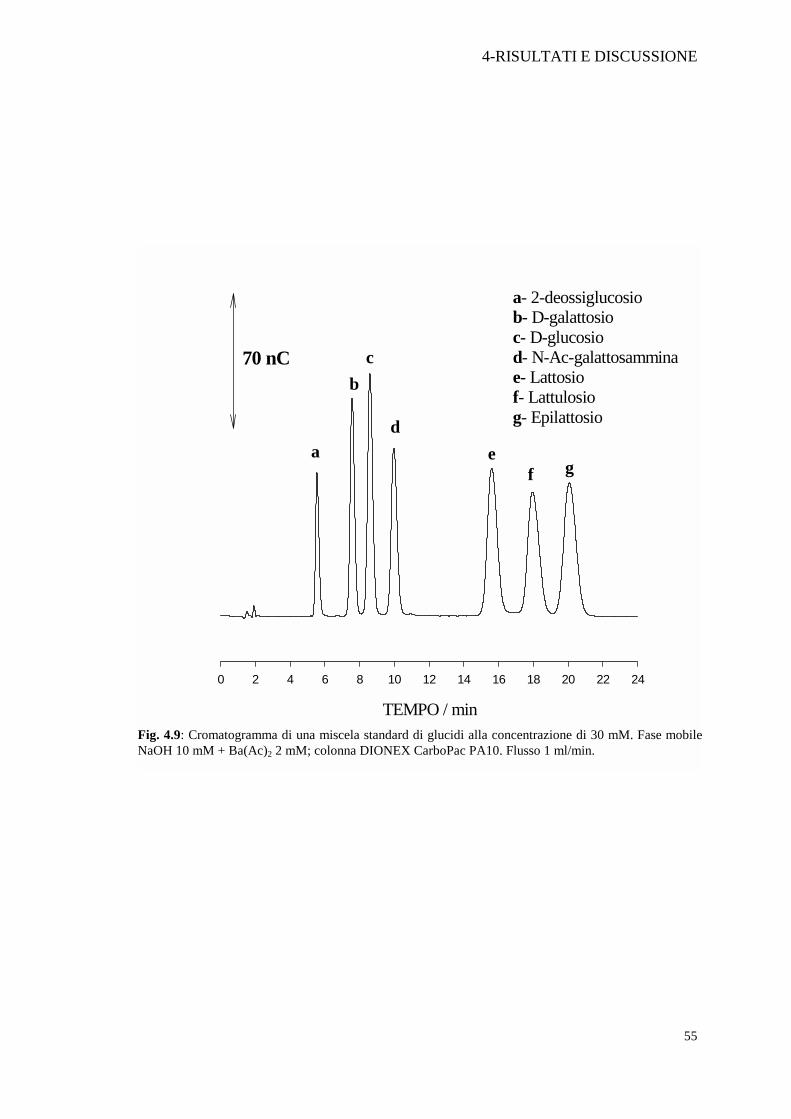
scelta migliore in quanto eluisce nei primi minuti di analisi (tR=5.46 min) e non
interferisce con nessuno degli analiti di interesse presenti nel latte. Dopo il trattamento
con Carrez, il confronto tra la concentrazione stimata del nostro standard interno e la
concentrazione effettiva, considerato il fattore di diluizione, ci ha permesso di
affermare che il trattamento di deproteinizzazione non altera la concentrazione dei
Fig. 4.9: Cromatogramma di una miscela standard di glucidi alla concentrazione di 30 mM. Fase mobileNaOH 10 mM + Ba(Ac)2 2 mM; colonna DIONEX CarboPac PA10. Flusso 1 ml/min.
4-RISULTATI E DISCUSSIONE
56
4.6. ANALISI DI UN CAMPIONE REALE: PROBLEMI LEGATI ALLE
CONCENTRAZIONI RELATIVE DI LATTOSIO E LATTULOSIO
PRESENTE NEI CAMPIONI REALI
Come già riportato nella parte introduttiva, il lattosio è il carboidrato più
rappresentativo presente nel latte, con un contenuto che si aggira tra il 4.7 e il 5.0%
(p/v). Il lattulosio è invece assente nel latte naturale e si forma per isomerizzazione
termica del lattosio in quantità pari circa 1/1000 in funzione della durata e del
trattamento termico. Le moderne tecnologie sono in grado di limitare al meglio il
danno termico provocato al latte durante la pastorizzazione e la sterilizzazione,
contenendo al livello più basso possibile la quantità di lattulosio formatosi. Il
lattulosio (4-O-β-D-galattopiranosil-D-fruttosio) è un disaccaride isomero del
lattosio le cui proprietà chimiche sono molto simili. Come riportato in Figura 4.9 la
loro separazione è possibile con buona risoluzione (Rs=2.18) nelle condizioni di fase
mobile ottimizzate e descritte in precedenza. Sfortunatamente, la risoluzione
peggiora sensibilmente nei campioni reali dove la concentrazione di lattosio sovrasta
di gran lunga quella del lattulosio. Questo risulta particolarmente evidente
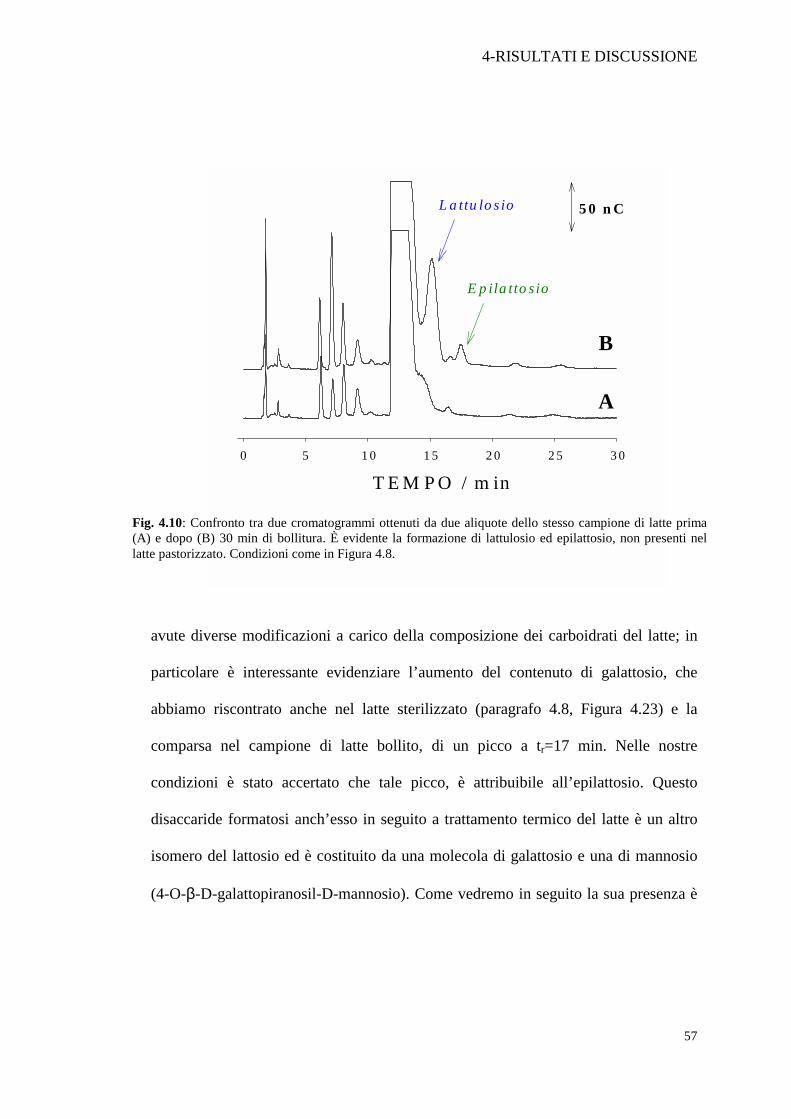
dall’osservazione della Figura 4.10. Come prova qualitativa è stato sottoposto un
campione di latte a bollitura per 30 minuti. Due aliquote del campione, di cui una
bollita, sono state pretrattate con i reagenti di Carrez, diluite 1:10, filtrate ed iniettate
in colonna per verificare la formazione di lattulosio. Come dimostrato in Figura 4.10,
la formazione di lattulosio in un campione di latte pastorizzato, sottoposto a circa
100°C per 30 minuti è chiaramente evidente. Durante il trattamento termico si sono
4-RISULTATI E DISCUSSIONE
57
avute diverse modificazioni a carico della composizione dei carboidrati del latte; in
particolare è interessante evidenziare l’aumento del contenuto di galattosio, che
abbiamo riscontrato anche nel latte sterilizzato (paragrafo 4.8, Figura 4.23) e la
comparsa nel campione di latte bollito, di un picco a tr=17 min. Nelle nostre
condizioni è stato accertato che tale picco, è attribuibile all’epilattosio. Questo
disaccaride formatosi anch’esso in seguito a trattamento termico del latte è un altro
isomero del lattosio ed è costituito da una molecola di galattosio e una di mannosio
(4-O-β-D-galattopiranosil-D-mannosio). Come vedremo in seguito la sua presenza è
T E M P O / m in0 5 1 0 1 5 2 0 2 5 3 0
5 0 n CL a ttu lo s io
E p ila tto s io
A
B
Fig. 4.10: Confronto tra due cromatogrammi ottenuti da due aliquote dello stesso campione di latte prima (A) e dopo (B) 30 min di bollitura. È evidente la formazione di lattulosio ed epilattosio, non presenti nel latte pastorizzato. Condizioni come in Figura 4.8.
4-RISULTATI E DISCUSSIONE
58
strettamente correlata a quella del lattulosio e quindi anch’esso può essere preso
come riferimento per stabilire l’entità del trattamento termico subito dal latte [29].
Dall’esame della Figura 4.10 si può notare che il picco del lattosio presenta
una codatura molto pronunciata. Questo fenomeno potrebbe essere attribuito ad un
avvelenamento della superficie dell’elettrodo di lavoro, che in presenza di elevate
quantità di lattosio non riesce, se non dopo alcuni secondi, a ripristinare le condizioni
iniziali. Parte del presente lavoro di tesi è stato finalizzato a minimizzare questo
problema agendo sostanzialmente su due fronti:
# Migliorare le condizioni di rivelazione modificando la forma d’onda
applicata all’elettrodo di lavoro;
# Migliorare ulteriormente la separazione tra lattosio e lattulosio agendo
sulla composizione della fase mobile, sulla colonna e sul flusso.
4-RISULTATI E DISCUSSIONE
59
4.7. STRATEGIE DI MIGLIORAMENTO DEI PARAMETRI DI
RIVELAZIONE
OTTIMIZZAZIONE DELLA FORMA D’ONDA POTENZIALE/TEMPO
Come già accennato in precedenza, nella rivelazione amperometrica pulsata i
parametri che possono essere ottimizzati sono i seguenti [34, 35, 36, 37]:
• potenziale di determinazione, il quale deve essere scelto a seconda della
classe dei composti da analizzare;
• potenziali di ossidazione e di riduzione che devono essere idonei ad
ottenere una adeguata pulizia della superficie elettrodica;
• l’ambiente di rivelazione, inteso come condizioni chimico-fisiche in cui
l’elettrodo deve svolgere l'ossidazione elettrocatalitica (pH della fase
mobile, temperatura, flusso ecc.).
Normalmente il potenziale di determinazione può essere scelto con l’ausilio
della voltammetria ciclica [20]. Come già anticipato nel nostro caso si è trattato di
trovare i potenziali e i tempi ottimali, in grado di fornire segnali elevati e ridotta
codatura del picco del lattosio. Per valutare i parametri della forma d’onda più adatti
per l’analisi dei campioni di latte, sono state fatte una serie di prove mirate
all’ottimizzazione delle singole variabili. Le impostazioni di base sono state tratte dal
lavoro di LaCourse e Johnson [18] sull’ottimizzazione della forma d’onda per
l’analisi dei carboidrati, utilizzando un elettrodo di lavoro d’oro e una fase mobile
NaOH 100 mM. I tempi e i potenziali sono riassunti nella Tabella 4.5.
4-RISULTATI E DISCUSSIONE
60
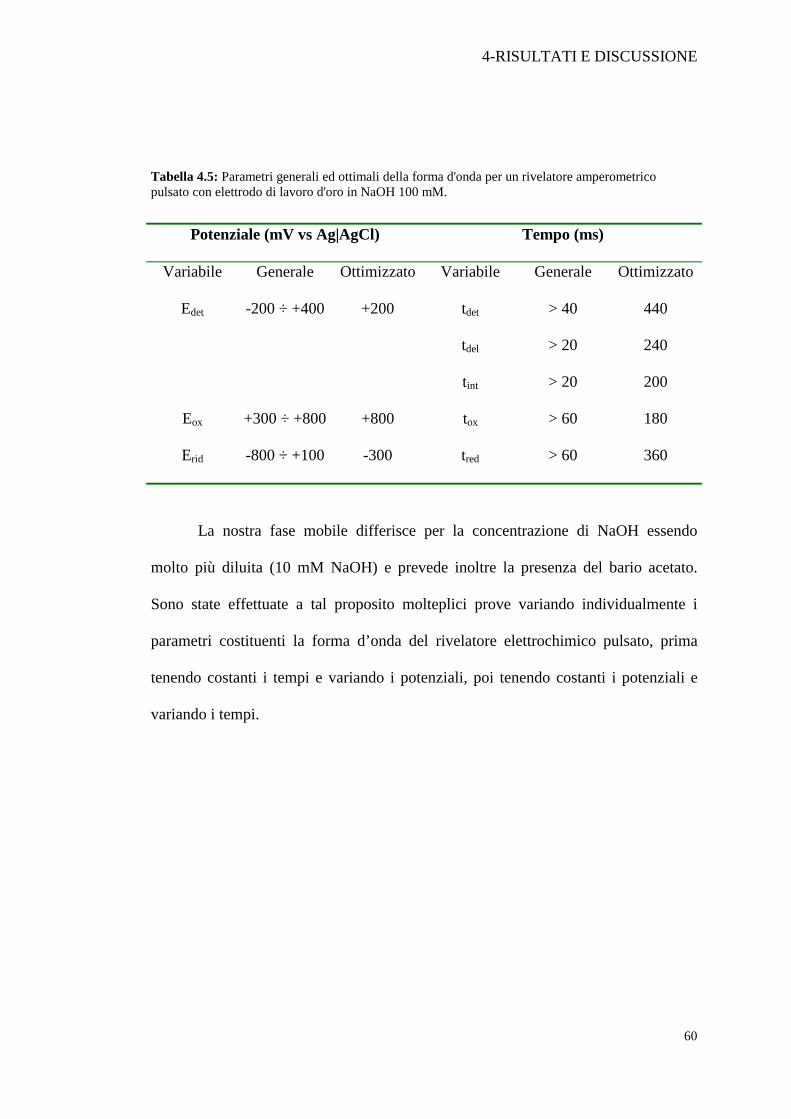
Tabella 4.5: Parametri generali ed ottimali della forma d'onda per un rivelatore amperometrico pulsato con elettrodo di lavoro d'oro in NaOH 100 mM.
Potenziale (mV vs Ag|AgCl) Tempo (ms)
Variabile Generale Ottimizzato Variabile Generale Ottimizzato
tdet > 40 440
tdel > 20 240
Edet -200 ÷ +400 +200
tint > 20 200
Eox +300 ÷ +800 +800 tox > 60 180
Erid -800 ÷ +100 -300 tred > 60 360
La nostra fase mobile differisce per la concentrazione di NaOH essendo
molto più diluita (10 mM NaOH) e prevede inoltre la presenza del bario acetato.
Sono state effettuate a tal proposito molteplici prove variando individualmente i
parametri costituenti la forma d’onda del rivelatore elettrochimico pulsato, prima
tenendo costanti i tempi e variando i potenziali, poi tenendo costanti i potenziali e
variando i tempi.
4-RISULTATI E DISCUSSIONE
61
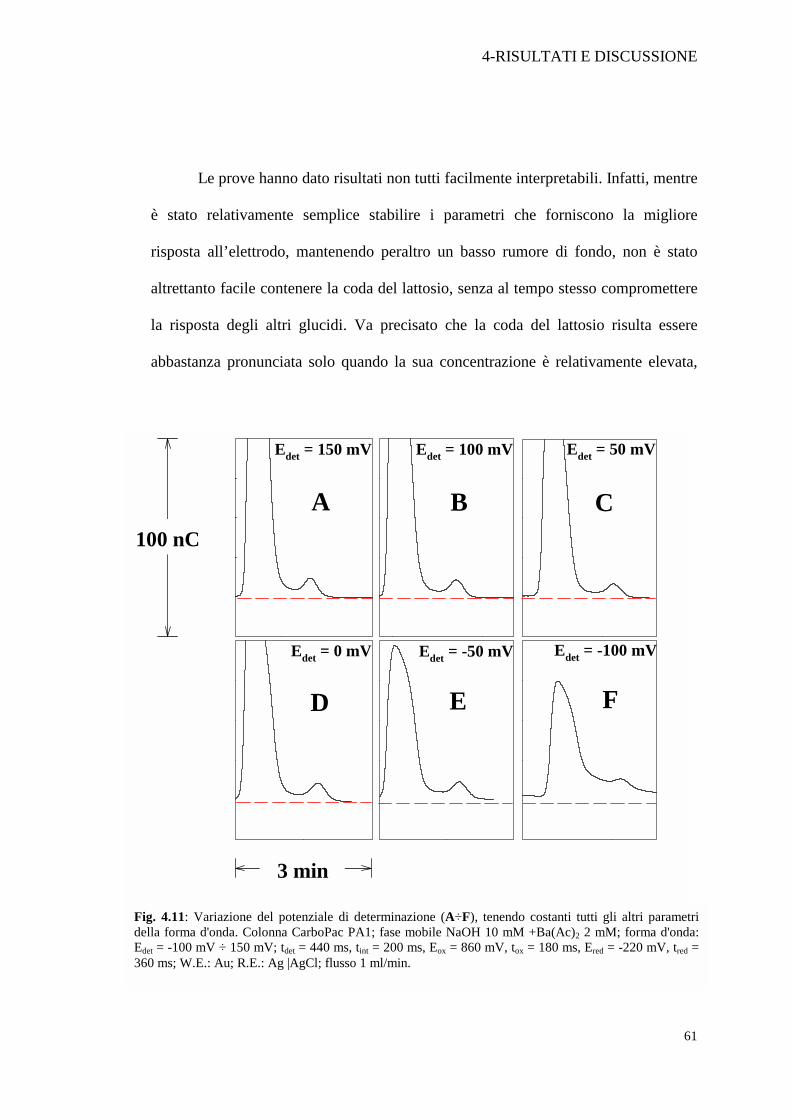
Le prove hanno dato risultati non tutti facilmente interpretabili. Infatti, mentre
è stato relativamente semplice stabilire i parametri che forniscono la migliore
risposta all’elettrodo, mantenendo peraltro un basso rumore di fondo, non è stato
altrettanto facile contenere la coda del lattosio, senza al tempo stesso compromettere
la risposta degli altri glucidi. Va precisato che la coda del lattosio risulta essere
abbastanza pronunciata solo quando la sua concentrazione è relativamente elevata,
Edet = 150 mV Edet = 100 mV Edet = 50 mV
Edet = 0 mV Edet = -50 mV Edet = -100 mV
A B C
D E F
3 min
100 nC
Fig. 4.11: Variazione del potenziale di determinazione (A÷F), tenendo costanti tutti gli altri parametri della forma d'onda. Colonna CarboPac PA1; fase mobile NaOH 10 mM +Ba(Ac)2 2 mM; forma d'onda: Edet = -100 mV ÷ 150 mV; tdet = 440 ms, tint = 200 ms, Eox = 860 mV, tox = 180 ms, Ered = -220 mV, tred = 360 ms; W.E.: Au; R.E.: Ag |AgCl; flusso 1 ml/min.
4-RISULTATI E DISCUSSIONE
62
come si verifica nei campioni di latte, seppur diluiti di un fattore compreso tra 1:10 e
1:100. Abbiamo inoltre potuto verificare che l’effetto di codatura del picco del lattosio
compare in maniera netta solo quando la concentrazione di soda in fase mobile è bassa
(es. 10-20 mM) mentre non si riscontra per concentrazioni 100-200 mM di NaOH. Si
confronti a tal proposito la Figura 4.1 a pag. 37 con la Figura 4.11 C.
Nell’intento di migliorare la rivelazione del lattosio si è anche tentato di
utilizzare una forma d’onda recentemente proposta da Rocklin e collaboratori [35]
per la determinazione dei carboidrati su un elettrodo d’oro. Uno dei parametri
importanti della rivelazione amperometrica pulsata è lo step di pulizia della
superficie elettrodica. Dato che l’attività elettrochimica dell’elettrodo d’oro in
ambiente alcalino può modificarsi in seguito all’adsorbimento dei prodotti di
ossidazione dei carboidrati, questo potrebbe essere uno dei motivi per cui si ha la
presenza della codatura al picco del lattosio. Nel lavoro di Rocklin e collaboratori lo
scopo della nuova forma d’onda era quello di minimizzare l’usura della superficie
elettrodica dovuta all’applicazione di potenziali di ossidazione per tempi
relativamente lunghi (tox=200 ms). Per diminuire questo effetto e
contemporaneamente avere una buona pulizia dell’elettrodo e stato proposto di
praticare un desorbimento catodico a potenziali molto negativi (Eclean=-2 V), che
favorirebbe una pulizia più rapida della superficie elettrodica, rispetto alla
tradizionale forma d’onda a tre potenziali e permetterebbe una rapida ripresa
dell’attività dell’elettrodo di lavoro. Nella Figura 4.12 sono confrontati i
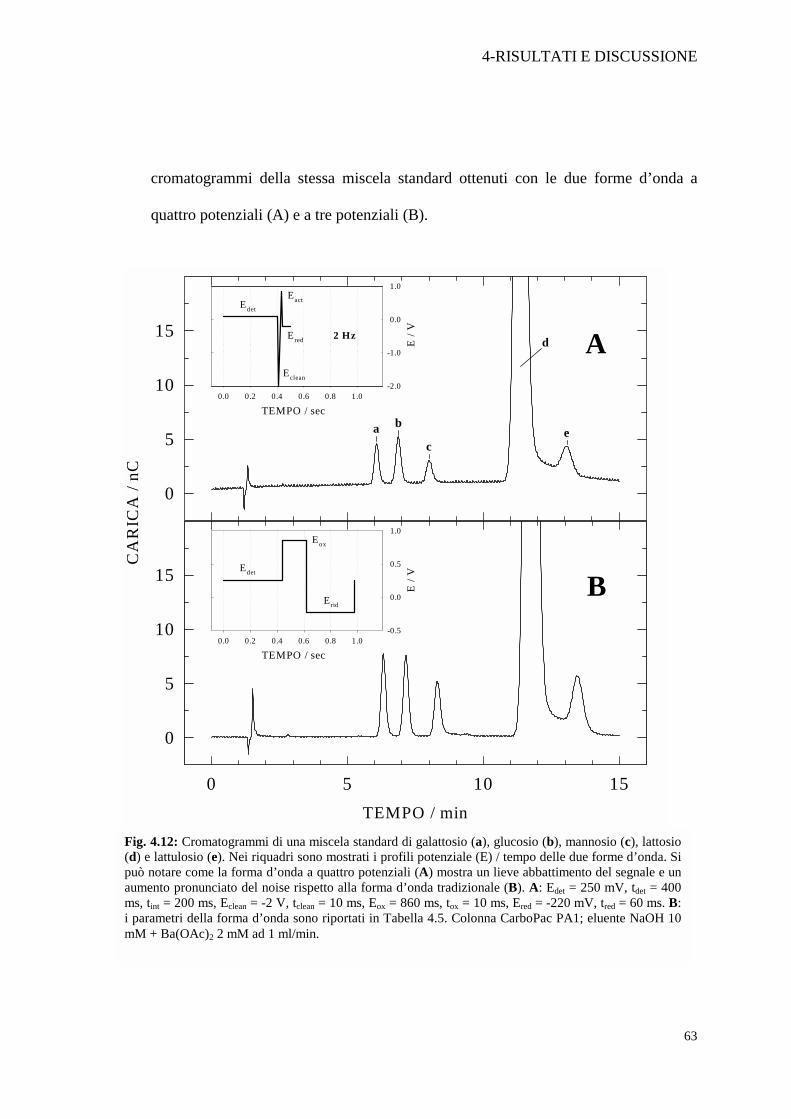
4-RISULTATI E DISCUSSIONE
63
cromatogrammi della stessa miscela standard ottenuti con le due forme d’onda a
quattro potenziali (A) e a tre potenziali (B).
TEMPO / min
0 5 10 15
CA
RIC
A /
nC
0
5
10
15
0
5
10
15
TEMPO / sec0.0 0.2 0.4 0.6 0.8 1.0
E /
V
-0.5
0.0
0.5
1.0
TEMPO / sec0.0 0.2 0.4 0.6 0.8 1.0
E /
V
-2.0
-1.0
0.0
1.0
a b
ce
d
Edet
Eclean
Eact
Ered 2 Hz
Edet
Eox
Erid
A
B
Fig. 4.12: Cromatogrammi di una miscela standard di galattosio (a), glucosio (b), mannosio (c), lattosio (d) e lattulosio (e). Nei riquadri sono mostrati i profili potenziale (E) / tempo delle due forme d’onda. Si può notare come la forma d’onda a quattro potenziali (A) mostra un lieve abbattimento del segnale e un aumento pronunciato del noise rispetto alla forma d’onda tradizionale (B). A: Edet = 250 mV, tdet = 400 ms, tint = 200 ms, Eclean = -2 V, tclean = 10 ms, Eox = 860 ms, tox = 10 ms, Ered = -220 mV, tred = 60 ms. B: i parametri della forma d’onda sono riportati in Tabella 4.5. Colonna CarboPac PA1; eluente NaOH 10 mM + Ba(OAc)2 2 mM ad 1 ml/min.
4-RISULTATI E DISCUSSIONE
64
Dall’esame della Figura 4.12 A si evince che la nuova forma d’onda non
migliora la codatura del lattosio; inoltre è evidente un notevole aumento del noise
della linea di base e una diminuizione del segnale di tutti i carboidrati. Questo ci ha
spinti ad utilizzare, per il proseguo del lavoro, la forma d’onda tradizionale a tre
potenziali, cercando di ottimizzare al meglio i singoli parametri.
Abbiamo potuto stabilire che i valori ottimali per quanto attiene i potenziali di
ossidazione e riduzione nelle nostre condizioni sono: Eox = +860 mV, Erid = -220 mV.
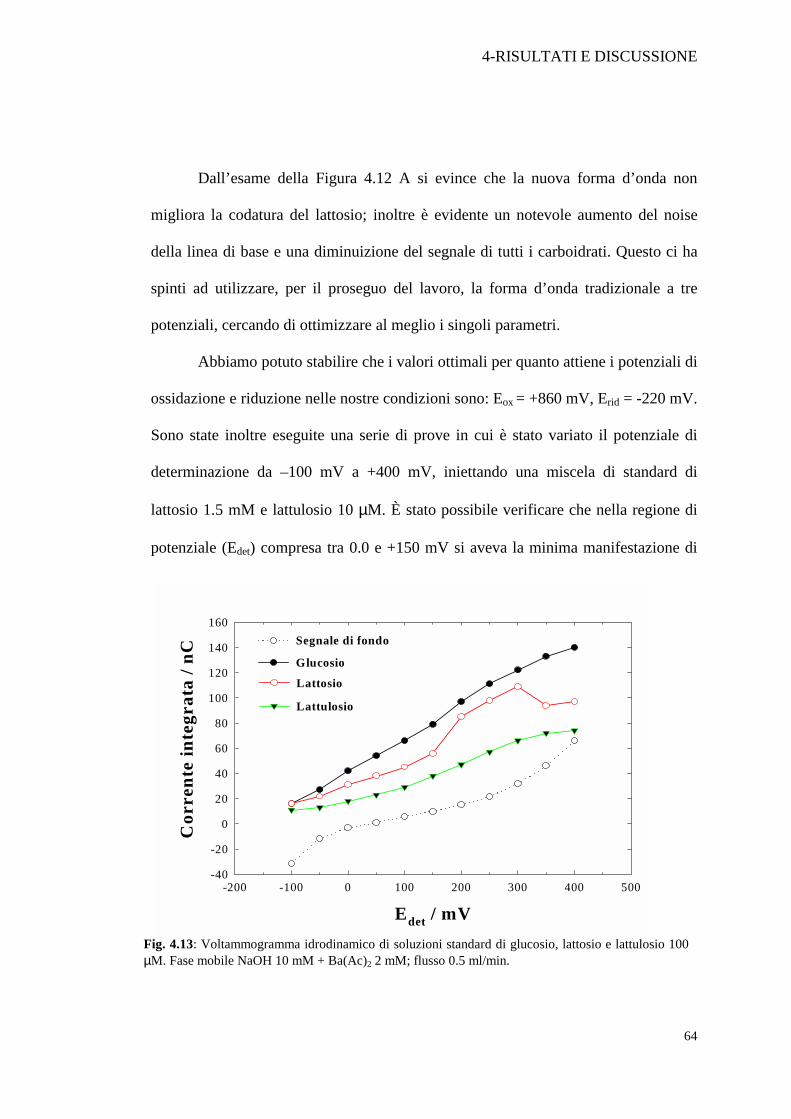
Sono state inoltre eseguite una serie di prove in cui è stato variato il potenziale di
determinazione da –100 mV a +400 mV, iniettando una miscela di standard di
lattosio 1.5 mM e lattulosio 10 µM. È stato possibile verificare che nella regione di
potenziale (Edet) compresa tra 0.0 e +150 mV si aveva la minima manifestazione di
Edet / mV-200 -100 0 100 200 300 400 500
Cor
rent
e in
tegr
ata
/ nC
-40
-20
0
20
40
60
80
100
120
140
160Segnale di fondo
Glucosio
Lattosio
Lattulosio
Fig. 4.13: Voltammogramma idrodinamico di soluzioni standard di glucosio, lattosio e lattulosio 100 µM. Fase mobile NaOH 10 mM + Ba(Ac)2 2 mM; flusso 0.5 ml/min.
4-RISULTATI E DISCUSSIONE
65
codatura del lattosio. In particolare si è preferito lavorare ad un potenziale di
determinazione di +50 mV, perché a tale valore il segnale di fondo è prossimo allo
zero e consente di ottenere il miglior rapporto segnale/rumore. (Figura 4.13). Nella
Tabella 4.6 sono riassunti tutti i parametri della forma d’onda da noi ottimizzata nelle
nostre condizioni di lavoro.
Tabella 4.6: Forma d'onda ottimizzata.
Potenziale mV Tempo/ms
Edet +50 440(totale) 200(integrazione)
Eox +860 180
Ered -220 360
AUMENTO DEL pH PER AGGIUNTA POST-COLONNA DI BASE
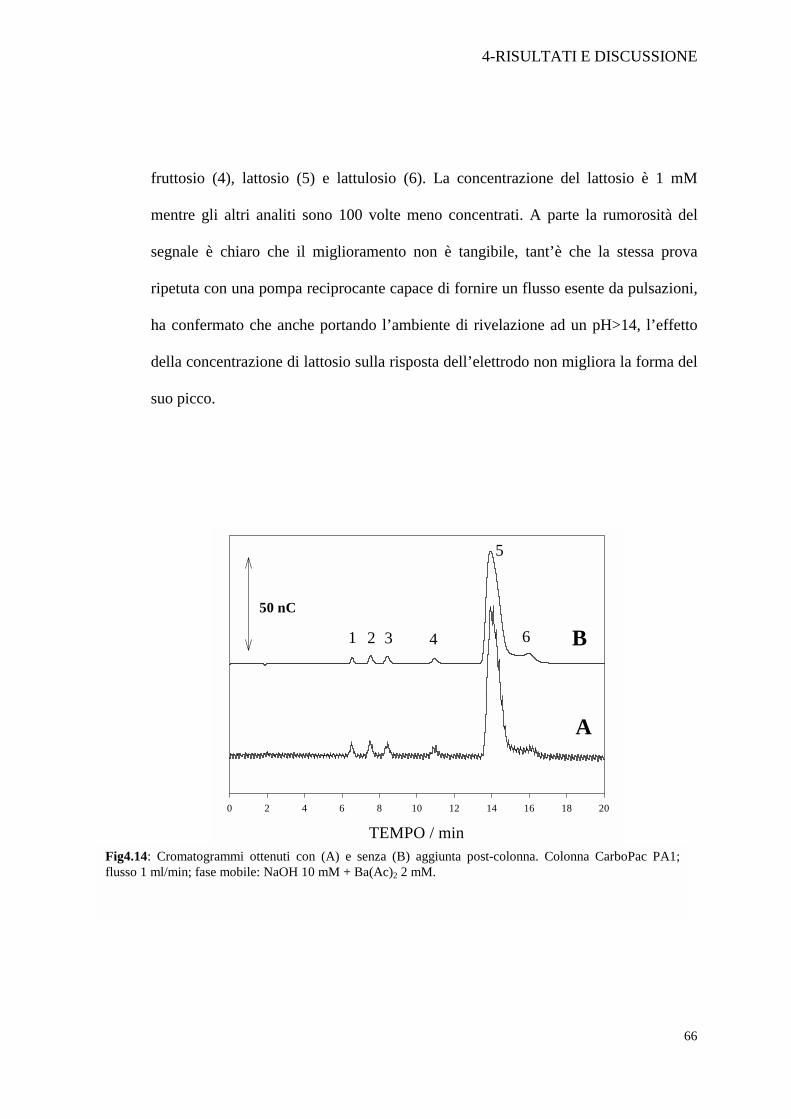
Dalla Figura 4.1 si nota che la coda del lattosio non è presente ad alte
concentrazioni di NaOH. Si è pensato allora di alcalinizzare la fase mobile
aggiungendo soda concentrata in post-colonna per migliorare la rivelazione senza
pregiudicare la separazione. La prima prova è stata fatta utilizzando una pompa
peristaltica con cui veniva pompata NaOH 200 mM ad un flusso di 1 ml/min, ma le
pulsazioni del flusso, come mostrato in Figura 4.14 A, provocavano un forte aumento
di rumorosità del segnale. La prova è stata effettuata in doppio, con e senza aggiunta
post-colonna, su uno standard contenente arabinosio (1), galattosio (2), glucosio (3),
4-RISULTATI E DISCUSSIONE
66
fruttosio (4), lattosio (5) e lattulosio (6). La concentrazione del lattosio è 1 mM
mentre gli altri analiti sono 100 volte meno concentrati. A parte la rumorosità del
segnale è chiaro che il miglioramento non è tangibile, tant’è che la stessa prova
ripetuta con una pompa reciprocante capace di fornire un flusso esente da pulsazioni,
ha confermato che anche portando l’ambiente di rivelazione ad un pH>14, l’effetto
della concentrazione di lattosio sulla risposta dell’elettrodo non migliora la forma del
suo picco.
TEMPO / min0 2 4 6 8 10 12 14 16 18 20
50 nC
A
B1 2 3 4
5
6
Fig4.14: Cromatogrammi ottenuti con (A) e senza (B) aggiunta post-colonna. Colonna CarboPac PA1; flusso 1 ml/min; fase mobile: NaOH 10 mM + Ba(Ac)2 2 mM.
4-RISULTATI E DISCUSSIONE
67
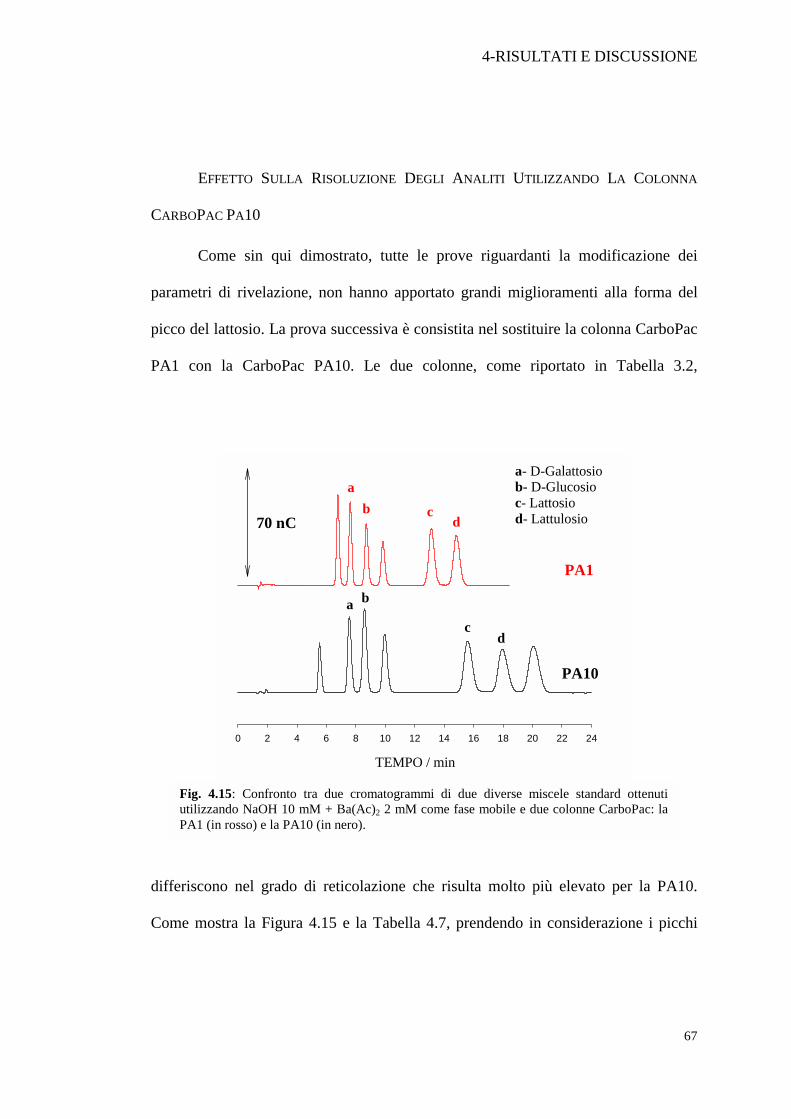
EFFETTO SULLA RISOLUZIONE DEGLI ANALITI UTILIZZANDO LA COLONNA
CARBOPAC PA10
Come sin qui dimostrato, tutte le prove riguardanti la modificazione dei
parametri di rivelazione, non hanno apportato grandi miglioramenti alla forma del
picco del lattosio. La prova successiva è consistita nel sostituire la colonna CarboPac
PA1 con la CarboPac PA10. Le due colonne, come riportato in Tabella 3.2,
differiscono nel grado di reticolazione che risulta molto più elevato per la PA10.
Come mostra la Figura 4.15 e la Tabella 4.7, prendendo in considerazione i picchi
Fig. 4.15: Confronto tra due cromatogrammi di due diverse miscele standard ottenuti utilizzando NaOH 10 mM + Ba(Ac)2 2 mM come fase mobile e due colonne CarboPac: la PA1 (in rosso) e la PA10 (in nero).
4-RISULTATI E DISCUSSIONE
68
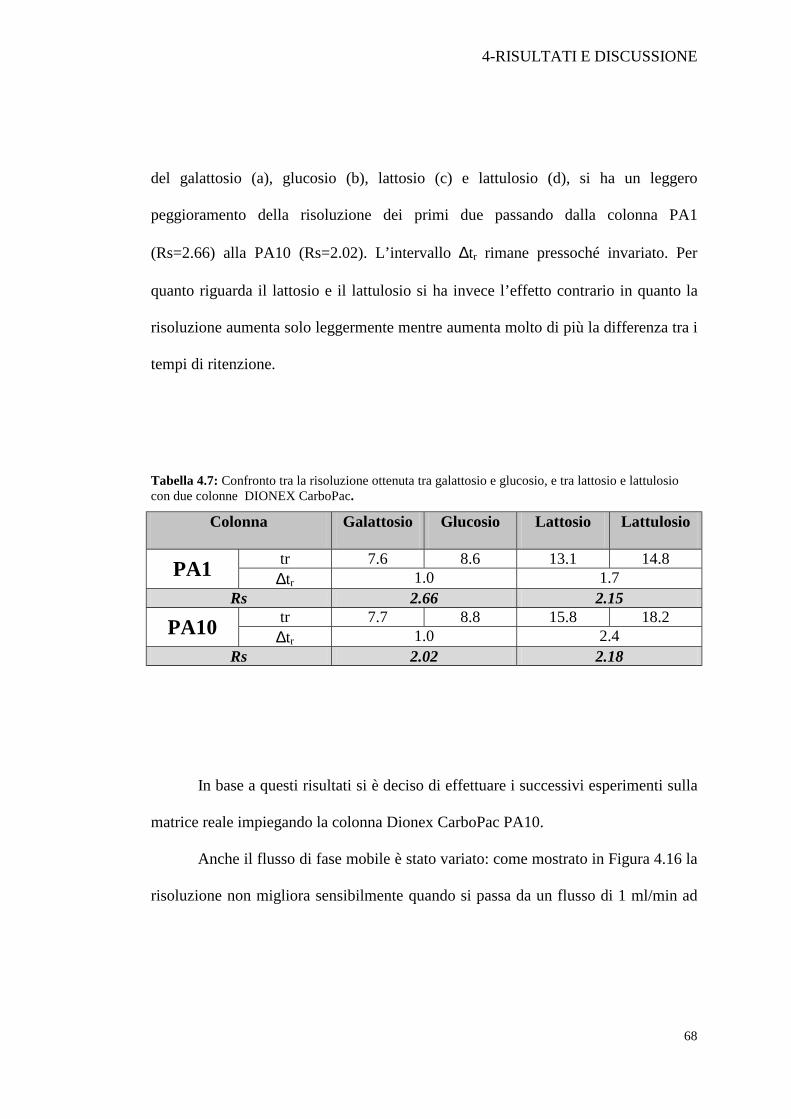
del galattosio (a), glucosio (b), lattosio (c) e lattulosio (d), si ha un leggero
peggioramento della risoluzione dei primi due passando dalla colonna PA1
(Rs=2.66) alla PA10 (Rs=2.02). L’intervallo ∆tr rimane pressoché invariato. Per
quanto riguarda il lattosio e il lattulosio si ha invece l’effetto contrario in quanto la
risoluzione aumenta solo leggermente mentre aumenta molto di più la differenza tra i
tempi di ritenzione.
Tabella 4.7: Confronto tra la risoluzione ottenuta tra galattosio e glucosio, e tra lattosio e lattulosio con due colonne DIONEX CarboPac.
In base a questi risultati si è deciso di effettuare i successivi esperimenti sulla
matrice reale impiegando la colonna Dionex CarboPac PA10.
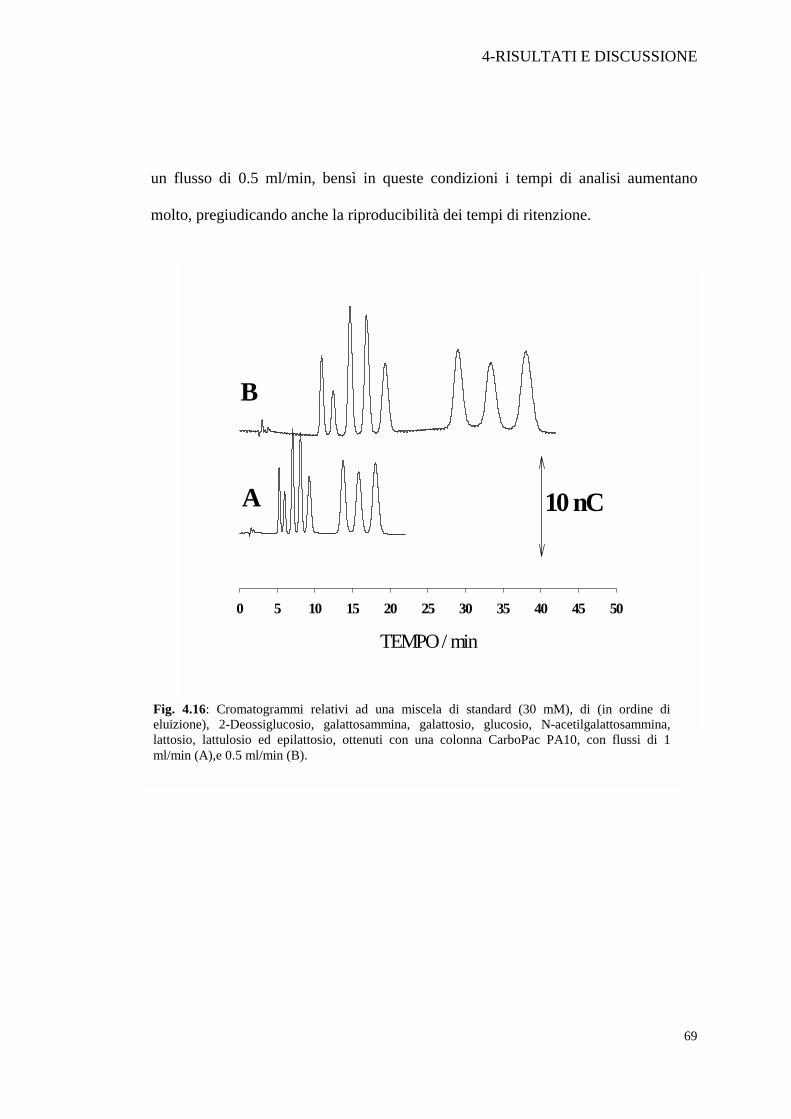
Anche il flusso di fase mobile è stato variato: come mostrato in Figura 4.16 la
risoluzione non migliora sensibilmente quando si passa da un flusso di 1 ml/min ad
4-RISULTATI E DISCUSSIONE
69
un flusso di 0.5 ml/min, bensì in queste condizioni i tempi di analisi aumentano
molto, pregiudicando anche la riproducibilità dei tempi di ritenzione.
TEMPO / min
0 5 10 15 20 25 30 35 40 45 50
A
B
10 nC
Fig. 4.16: Cromatogrammi relativi ad una miscela di standard (30 mM), di (in ordine dieluizione), 2-Deossiglucosio, galattosammina, galattosio, glucosio, N-acetilgalattosammina,lattosio, lattulosio ed epilattosio, ottenuti con una colonna CarboPac PA10, con flussi di 1ml/min (A),e 0.5 ml/min (B).
4-RISULTATI E DISCUSSIONE
70
C A
R I
C A
/
nC
0
20
40
60
PA 1
∆∆∆∆t = 5 min
0
20
40
60
PA 10
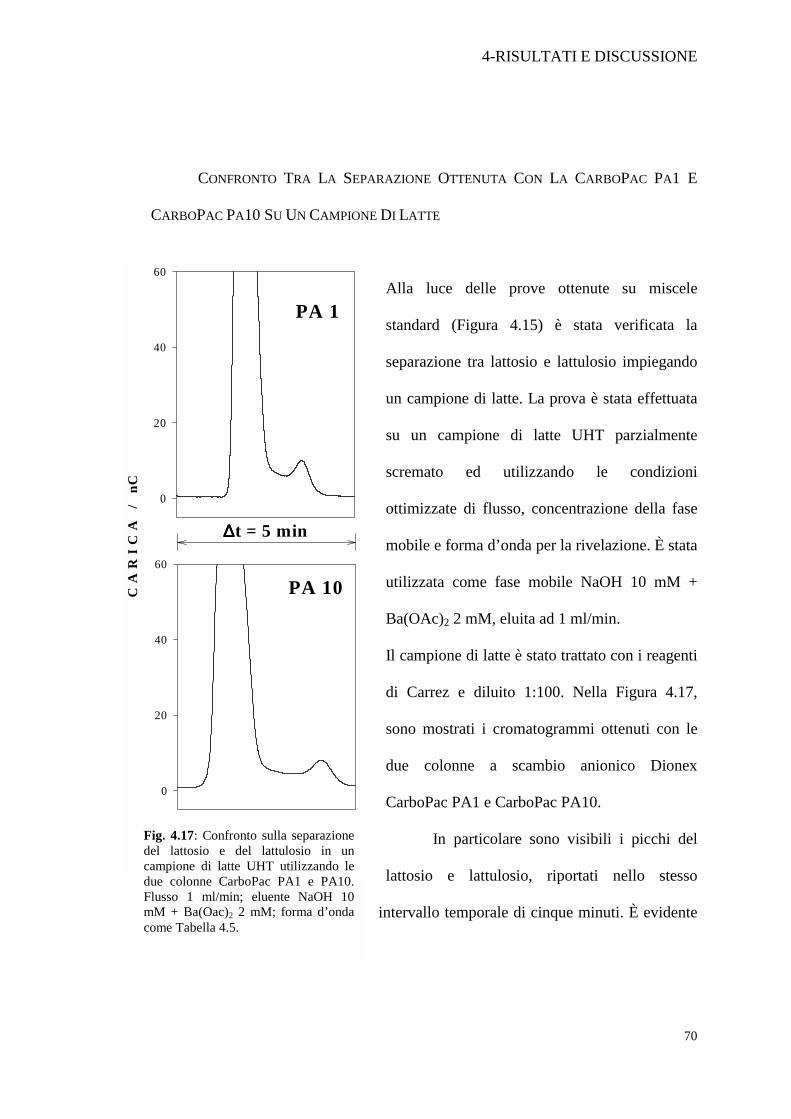
Fig. 4.17: Confronto sulla separazione del lattosio e del lattulosio in un campione di latte UHT utilizzando le due colonne CarboPac PA1 e PA10. Flusso 1 ml/min; eluente NaOH 10 mM + Ba(Oac)2 2 mM; forma d’onda come Tabella 4.5.
CONFRONTO TRA LA SEPARAZIONE OTTENUTA CON LA CARBOPAC PA1 E
CARBOPAC PA10 SU UN CAMPIONE DI LATTE
Alla luce delle prove ottenute su miscele
standard (Figura 4.15) è stata verificata la
separazione tra lattosio e lattulosio impiegando
un campione di latte. La prova è stata effettuata
su un campione di latte UHT parzialmente
scremato ed utilizzando le condizioni
ottimizzate di flusso, concentrazione della fase
mobile e forma d’onda per la rivelazione. È stata
utilizzata come fase mobile NaOH 10 mM +
Ba(OAc)2 2 mM, eluita ad 1 ml/min.
Il campione di latte è stato trattato con i reagenti
di Carrez e diluito 1:100. Nella Figura 4.17,
sono mostrati i cromatogrammi ottenuti con le
due colonne a scambio anionico Dionex
CarboPac PA1 e CarboPac PA10.
In particolare sono visibili i picchi del
lattosio e lattulosio, riportati nello stesso
intervallo temporale di cinque minuti. È evidente
4-RISULTATI E DISCUSSIONE
71
come la CarboPac PA10 permetta una migliore separazione e di conseguenza una
migliore stima della quantità di lattulosio. Da sottolineare come il picco del lattosio,
la cui intensità è in entrambi i casi fuori dal range di risposta lineare, risulti più
“allargato” usando la PA10 piuttosto che la PA1. Questo fenomeno è probabilmente
dovuto al maggiore tempo di permanenza nella colonna CarboPac PA10. Ad ogni
modo, anche se la codatura del picco relativo al lattosio è comunque presente, questo
non impedisce la determinazione del lattulosio con sufficiente accuratezza e buona
sensibilità.
INFLUENZA DELLO Zn2+
Un ultima prova mirata a migliorare la separazione tra lattosio e lattulosio è
consistita nel valutare l’effetto dell’aggiunta di zinco acetato alla fase mobile. In un
recente lavoro [38] è stata riportata la separazione del lattulosio contenuto in un
campione di latte UHT, utilizzando la colonna CarboPac PA1 ed una eluizione in
gradiente con NaOH 10 mM e Zn (OAc)2 0.5 mM. Abbiamo pertanto voluto valutare
l’impiego dello Zn(OAc)2 in concentrazioni di 0.2, 0.5 e 1 mM. Non sono state
utilizzate concentrazioni più elevate per la bassa solubilità dello Zn(OH)2 in
soluzione alcalina (Kps=3×10-16 in acqua a 25°C). Questo significa altresì che lo ione
zinco non è efficace quanto il bario e lo stronzio per la precipitazione del carbonato
presente in soluzione, sebbene il Kps dello ZnCO3 sia pari a 1×10-10. A tal proposito
si è tentato di modificare la fase mobile alcalina con miscele a diversa
concentrazione di zinco e bario acetato, per cercare di sfruttare la proprietà del bario
4-RISULTATI E DISCUSSIONE
72
nella precipitazione degli ioni carbonato e nello stesso tempo valutare l’efficacia dello zinco
sulla separazione tra lattosio e lattulosio. Nella Figura 4.18 sono poste a confronto due
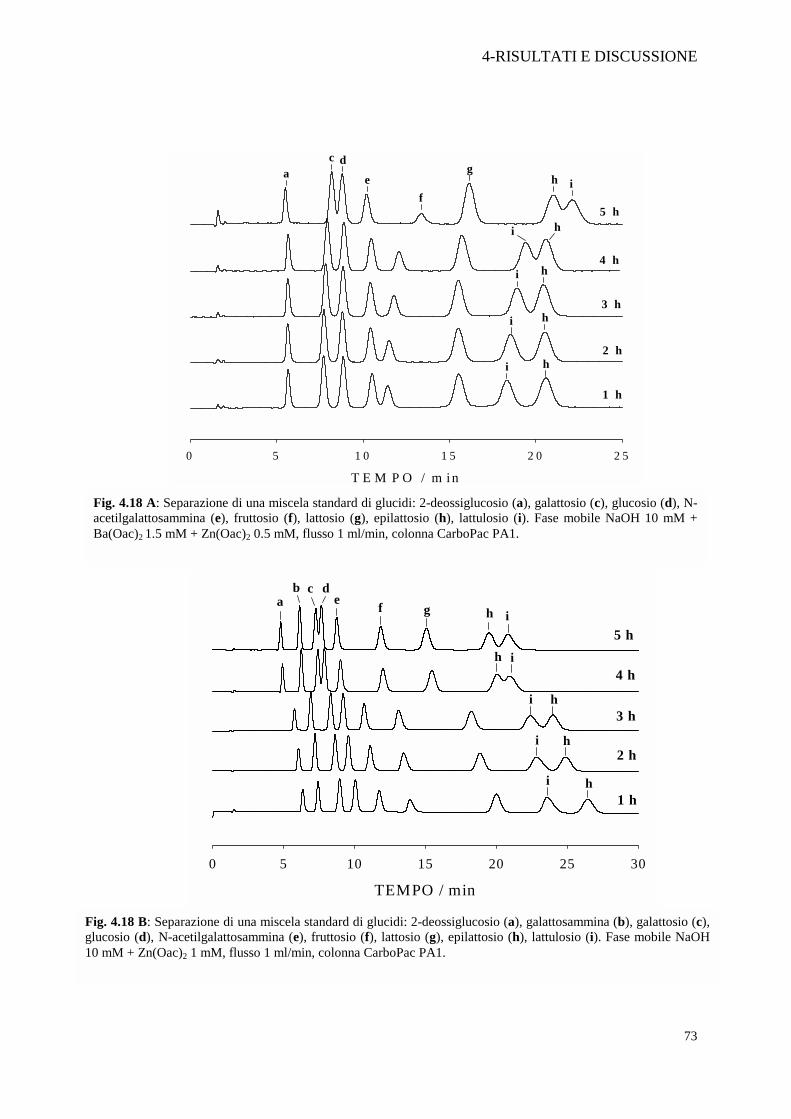
serie di cromatogrammi entrambi effettuati con la colonna CarboPac PA1, eluite con
NaOH 10 mM + Ba(OAc)2 1,5 mM + Zn(OAc)2 0,5 mM in un caso (Figura 4.18 A) e
NaOH 10 mM + Zn(OAc)2 1 mM nell’altro (Figura 4.18 B). Le prove hanno confermato
che l’effetto sulla riproducibilità dei tempi di ritenzione è da attribuire al bario; i picchi del 2-
deossiglucosio (a), del glucosio (d) e del mannosio (e), in cinque ore di lavoro, non mostrano
alcuna anticipazione dei tempi di ritenzione in presenza di Ba(OAc)2 1.5 mM (Figura 4.18
A). L’aggiunta dello zinco in fase mobile ha apportato un lento condizionamento della
colonna in entrambi i casi, rendendo inaffidabili i tempi di ritenzione di quasi tutti i
carboidrati. Lo stesso effetto non è riportato nel lavoro di Prodolliet [49] in quanto si
trattava di una separazione condotta in gradiente con colonna rigenerata dopo ogni
eluizione. Nel nostro caso si è osservato una finale coeluizione sia di galattosio e glucosio sia
di lattulosio ed epilattosio. Nelle Figure 4.18 A e B è evidente come col passare delle ore si
sovrappongono i picchi del galattosio e glucosio, con inversione dell’ordine di eluizione tra
lattulosio ed epilattosio.
L’effetto positivo sulla separazione del lattulosio, anche in presenza di elevate
quantità di lattulosio quindi, non può essere sfruttato nelle nostre condizioni sperimentali in
quanto per ottenere separazioni riproducibili si è costretti a rigenerare la colonna
dopo ogni corsa cromatografica.
4-RISULTATI E DISCUSSIONE
73
TEMPO / min
0 5 10 15 20 25 30
ab c d
e f g h i
h i
hi
hi
i h1 h
2 h
3 h
4 h
5 h
Fig. 4.18 B: Separazione di una miscela standard di glucidi: 2-deossiglucosio (a), galattosammina (b), galattosio (c), glucosio (d), N-acetilgalattosammina (e), fruttosio (f), lattosio (g), epilattosio (h), lattulosio (i). Fase mobile NaOH 10 mM + Zn(Oac)2 1 mM, flusso 1 ml/min, colonna CarboPac PA1.
Fig. 4.18 A: Separazione di una miscela standard di glucidi: 2-deossiglucosio (a), galattosio (c), glucosio (d), N-acetilgalattosammina (e), fruttosio (f), lattosio (g), epilattosio (h), lattulosio (i). Fase mobile NaOH 10 mM + Ba(Oac)2 1.5 mM + Zn(Oac)2 0.5 mM, flusso 1 ml/min, colonna CarboPac PA1.
T E M P O / m i n
0 5 1 0 1 5 2 0 2 5
1 h
2 h
3 h
4 h
5 h
ac d
ef
gih
i h
i h
i h
i h
4-RISULTATI E DISCUSSIONE
74
4.8. ANALISI QUANTITATIVA
Una volta definite tutte le condizioni sperimentali ottimali per eseguire la separazione
dei glucidi presenti in un campione di latte, si è passati alla fase applicativa del metodo. Le
condizioni ottimali prevedono una colonna DIONEX CarboPac PA10, una fase mobile NaOH
10 mM modificata con l’aggiunta di Ba(OAc)2 2 mM, un flusso di 1 ml/min, ed una forma
d’onda come da Tabella 4.6.
L’analisi quantitativa è stata mirata soprattutto alla determinazione del lattulosio, ma
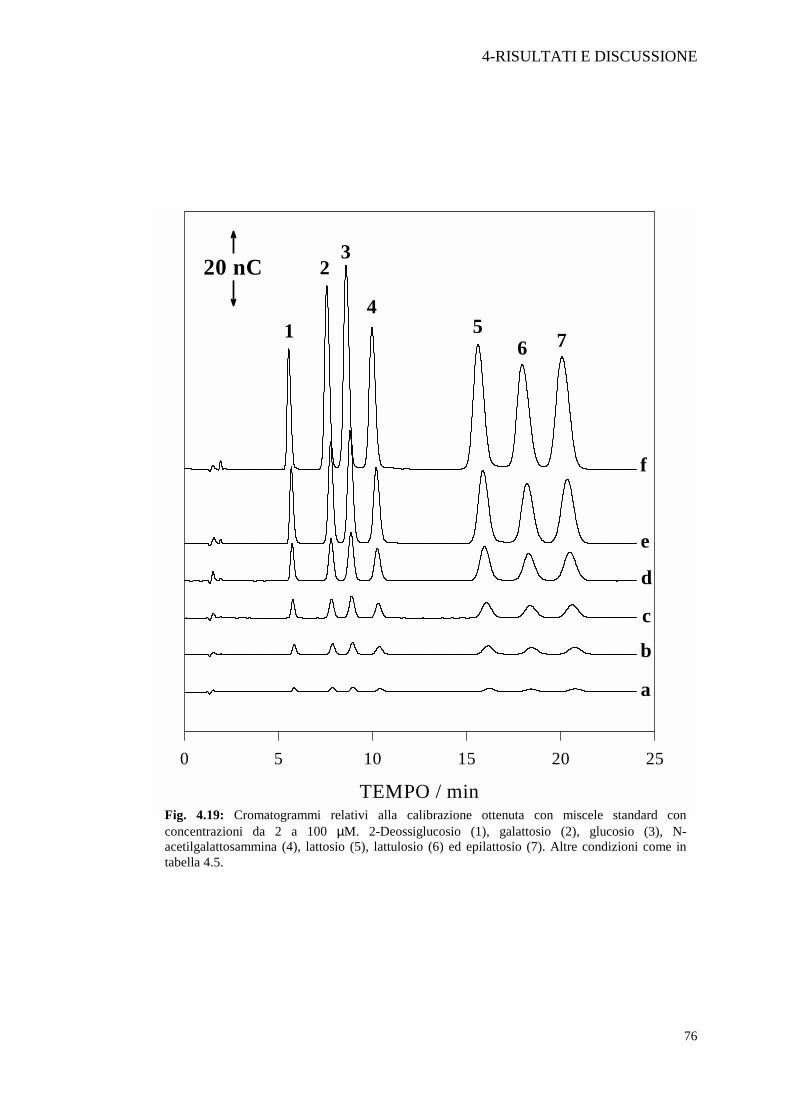
diversi glucidi minori sono stati quantificati. Per ognuno di tali composti è stata costruita la
curva di calibrazione, iniettando miscele standard a diverse concentrazioni e misurando
l’area corrispondente dei picchi cromatografici. Alcuni cromatogrammi sono mostrati
nella Figura 4.20. Per tutti i componenti le concentrazioni sono state comprese tra 2 (a)
e 100 (f) µM. Tutti gli analiti mostrano un buon coefficiente di correlazione (r). La
deviazione standard (SD) della pendenza e dell’intercetta è stata stimata ad un livello di
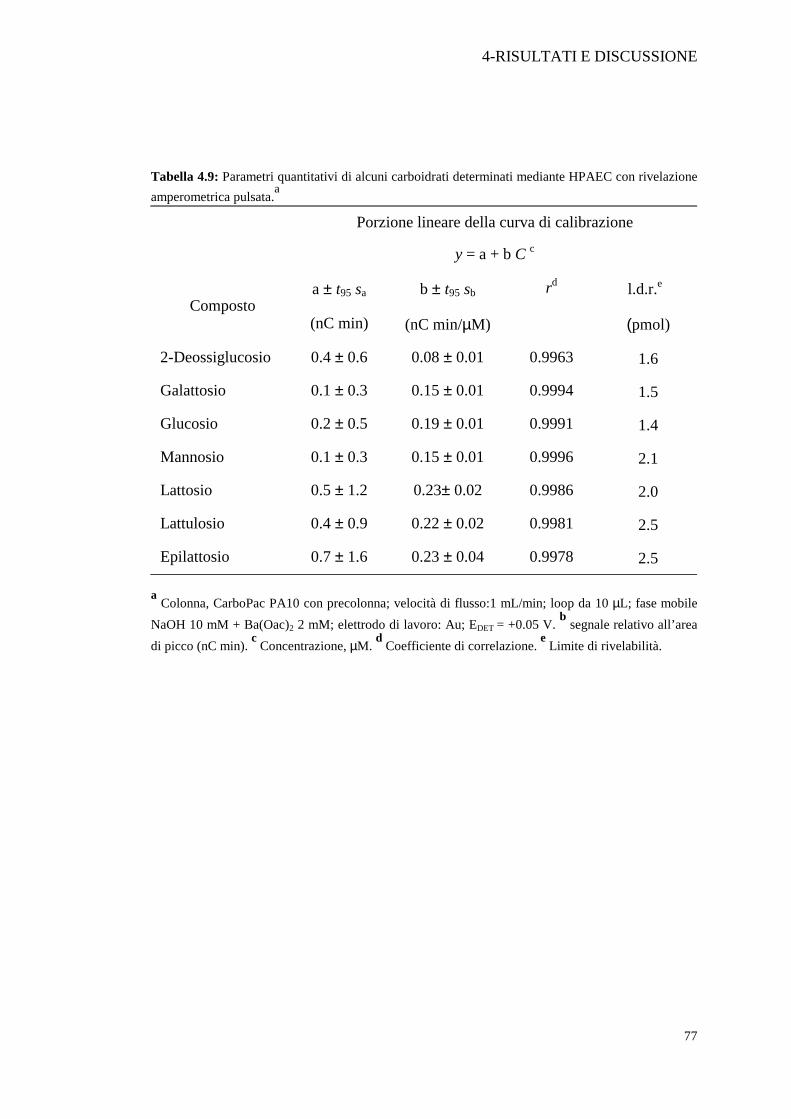
fiducia del 95%. Nella Tabella 4.9 sono mostrati i parametri di calibrazione ottenuti in
condizioni cromatografiche ottimizzate. Il limite di rivelabilità dei composti è stato calcolato
valutando il minimo segnale integrabile, assunto come tre volte il valore medio delle
fluttuazioni statistiche del segnale della linea di base.
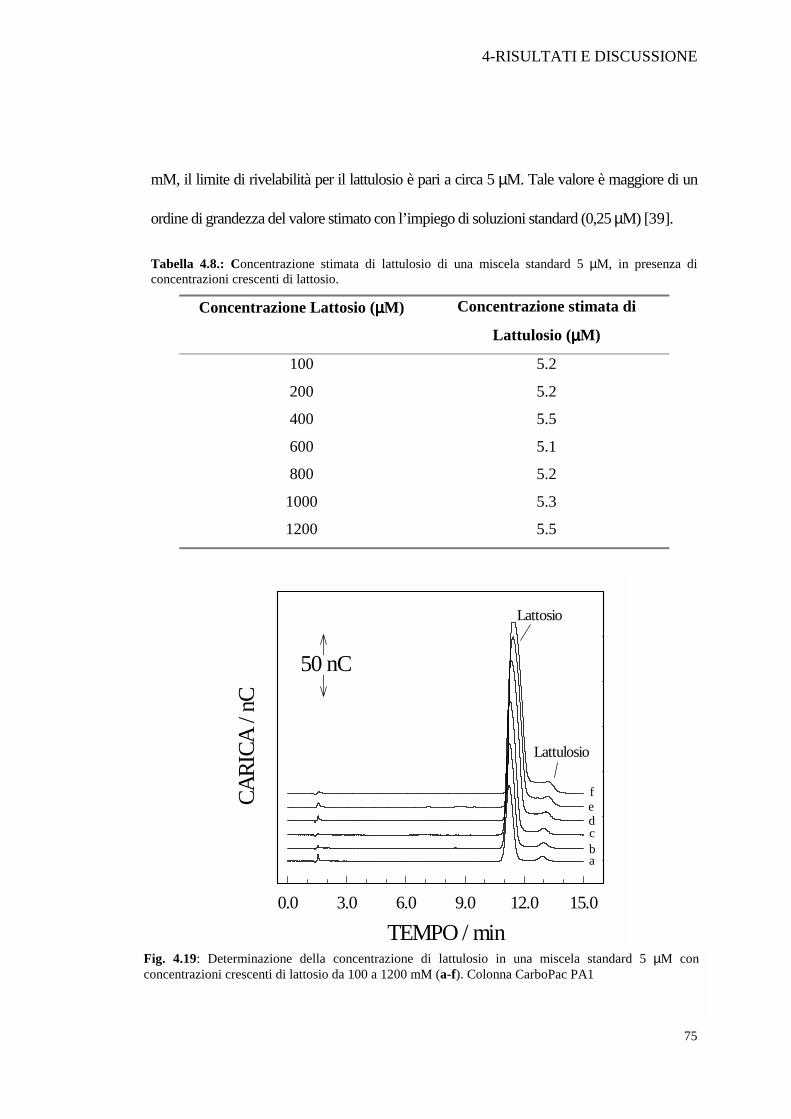
Una prima prova è stata mirata alla determinazione del lattulosio in presenza di diverse
concentrazioni di lattosio. A diverse aliquote di una soluzione standard di lattulosio 5 µM si
sono aggiunte quantità crescenti di lattosio, da 100 a 1200 µM. La concentrazione di lattulosio
è stata determinata mediante l’integrazione dell’area del picco. Le prove hanno mostrato
(Tabella 4.8 e Figura 4.19) che fino a quando la concentrazione di lattosio è inferiore a 1.2
4-RISULTATI E DISCUSSIONE
75
mM, il limite di rivelabilità per il lattulosio è pari a circa 5 µM. Tale valore è maggiore di un
ordine di grandezza del valore stimato con l’impiego di soluzioni standard (0,25 µM) [39].
Tabella 4.8.: Concentrazione stimata di lattulosio di una miscela standard 5 µM, in presenza di concentrazioni crescenti di lattosio.
Concentrazione Lattosio (µµµµM) Concentrazione stimata di
Lattulosio (µµµµM)
100 5.2
200 5.2
400 5.5
600 5.1
800 5.2
1000 5.3
1200 5.5
TEMPO / min0.0 3.0 6.0 9.0 12.0 15.0
CARI
CA /
nC
Lattulosio
Lattosio
abcd
50 nC
ef
Fig. 4.19: Determinazione della concentrazione di lattulosio in una miscela standard 5 µM con concentrazioni crescenti di lattosio da 100 a 1200 mM (a-f). Colonna CarboPac PA1
4-RISULTATI E DISCUSSIONE
76
TEMPO / min
0 5 10 15 20 25
a
b
c
d
e
f
20 nC
1
23
45
6 7
Fig. 4.19: Cromatogrammi relativi alla calibrazione ottenuta con miscele standard con concentrazioni da 2 a 100 µM. 2-Deossiglucosio (1), galattosio (2), glucosio (3), N-acetilgalattosammina (4), lattosio (5), lattulosio (6) ed epilattosio (7). Altre condizioni come in tabella 4.5.
4-RISULTATI E DISCUSSIONE
77
Tabella 4.9: Parametri quantitativi di alcuni carboidrati determinati mediante HPAEC con rivelazione amperometrica pulsata.a
Porzione lineare della curva di calibrazione
y = a + b C c
Composto a ± t95 sa
(nC min)
b ± t95 sb
(nC min/µM)
rd l.d.r.e
(pmol)
2-Deossiglucosio 0.4 ± 0.6 0.08 ± 0.01 0.9963 1.6
Galattosio 0.1 ± 0.3 0.15 ± 0.01 0.9994 1.5
Glucosio 0.2 ± 0.5 0.19 ± 0.01 0.9991 1.4
Mannosio 0.1 ± 0.3 0.15 ± 0.01 0.9996 2.1
Lattosio 0.5 ± 1.2 0.23± 0.02 0.9986 2.0
Lattulosio 0.4 ± 0.9 0.22 ± 0.02 0.9981 2.5
Epilattosio 0.7 ± 1.6 0.23 ± 0.04 0.9978 2.5
a Colonna, CarboPac PA10 con precolonna; velocità di flusso:1 mL/min; loop da 10 µL; fase mobile NaOH 10 mM + Ba(Oac)2 2 mM; elettrodo di lavoro: Au; EDET = +0.05 V. b segnale relativo all’area di picco (nC min). c Concentrazione, µM. d Coefficiente di correlazione. e Limite di rivelabilità.
4-RISULTATI E DISCUSSIONE
78
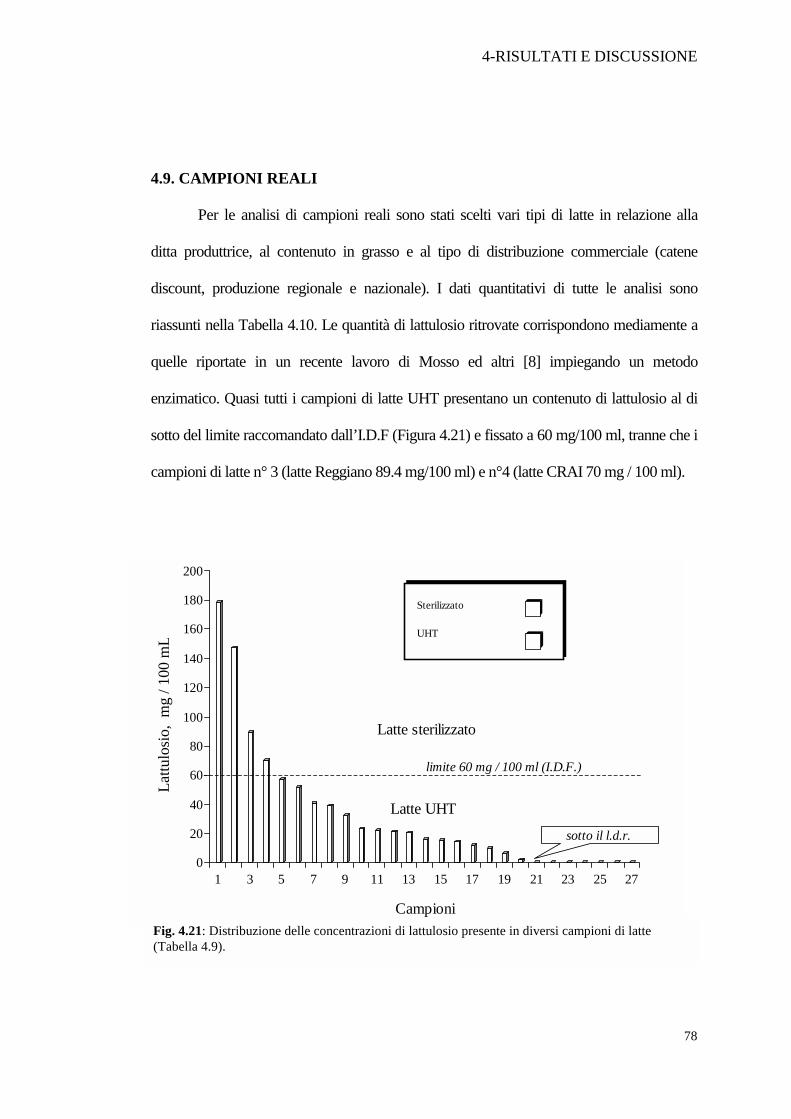
4.9. CAMPIONI REALI
Per le analisi di campioni reali sono stati scelti vari tipi di latte in relazione alla
ditta produttrice, al contenuto in grasso e al tipo di distribuzione commerciale (catene
discount, produzione regionale e nazionale). I dati quantitativi di tutte le analisi sono
riassunti nella Tabella 4.10. Le quantità di lattulosio ritrovate corrispondono mediamente a
quelle riportate in un recente lavoro di Mosso ed altri [8] impiegando un metodo
enzimatico. Quasi tutti i campioni di latte UHT presentano un contenuto di lattulosio al di
sotto del limite raccomandato dall’I.D.F (Figura 4.21) e fissato a 60 mg/100 ml, tranne che i
campioni di latte n° 3 (latte Reggiano 89.4 mg/100 ml) e n°4 (latte CRAI 70 mg / 100 ml).
0
20
40
60
80
100
120
140
160
180
200
Lattu
losi
o, m
g / 1
00 m
L
1 3 5 7 9 11 13 15 17 19 21 23 25 27
Campioni
Latte sterilizzato
Latte UHT
Sterilizzato
UHT
sotto il l.d.r.
limite 60 mg / 100 ml (I.D.F.)
Fig. 4.21: Distribuzione delle concentrazioni di lattulosio presente in diversi campioni di latte (Tabella 4.9).
4-RISULTATI E DISCUSSIONE
79
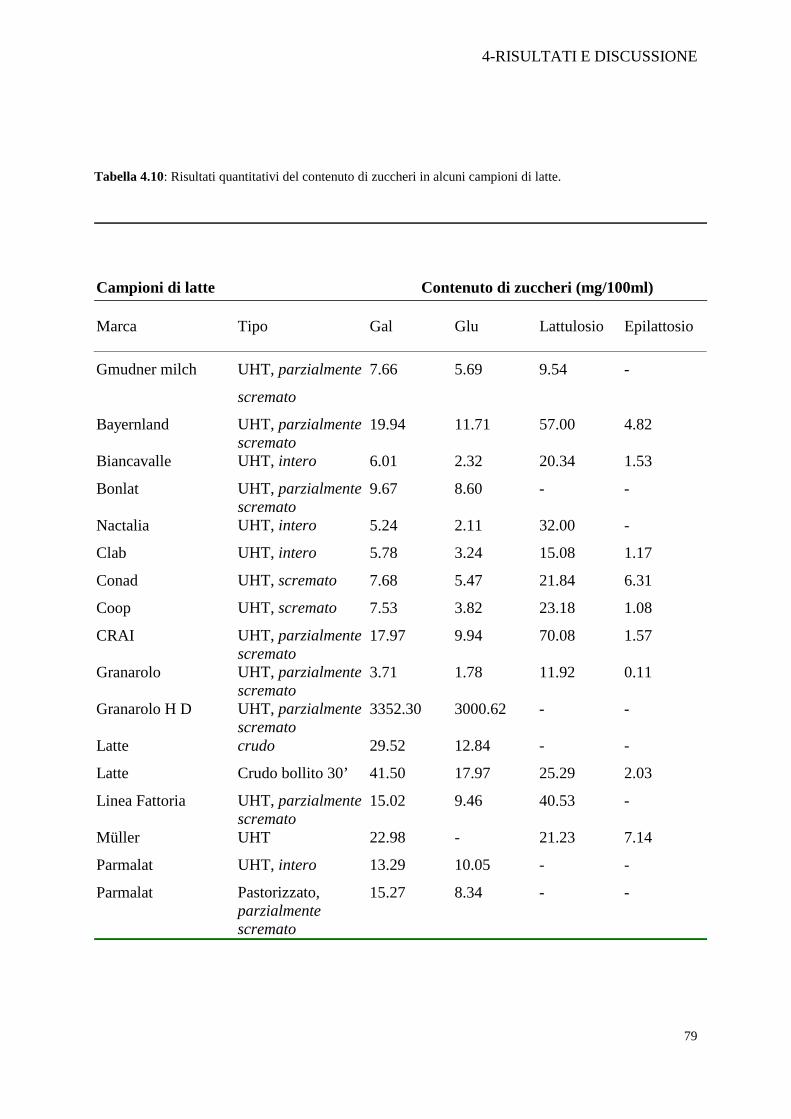
Tabella 4.10: Risultati quantitativi del contenuto di zuccheri in alcuni campioni di latte.
Campioni di latte
Contenuto di zuccheri (mg/100ml)
Marca Tipo Gal Glu Lattulosio Epilattosio
Gmudner milch UHT, parzialmente
scremato
7.66 5.69 9.54 -
Bayernland UHT, parzialmente scremato
19.94 11.71 57.00 4.82
Biancavalle UHT, intero 6.01 2.32 20.34 1.53
Bonlat UHT, parzialmente scremato
9.67 8.60 - -
Nactalia UHT, intero 5.24 2.11 32.00 -
Clab UHT, intero 5.78 3.24 15.08 1.17
Conad UHT, scremato 7.68 5.47 21.84 6.31
Coop UHT, scremato 7.53 3.82 23.18 1.08
CRAI UHT, parzialmente scremato
17.97 9.94 70.08 1.57
Granarolo UHT, parzialmente scremato
3.71 1.78 11.92 0.11
Granarolo H D UHT, parzialmente scremato
3352.30 3000.62 - -
Latte crudo 29.52 12.84 - -
Latte Crudo bollito 30’ 41.50 17.97 25.29 2.03
Linea Fattoria UHT, parzialmente scremato
15.02 9.46 40.53 -
Müller UHT 22.98 - 21.23 7.14
Parmalat UHT, intero 13.29 10.05 - -
Parmalat Pastorizzato, parzialmente scremato
15.27 8.34 - -
4-RISULTATI E DISCUSSIONE
80
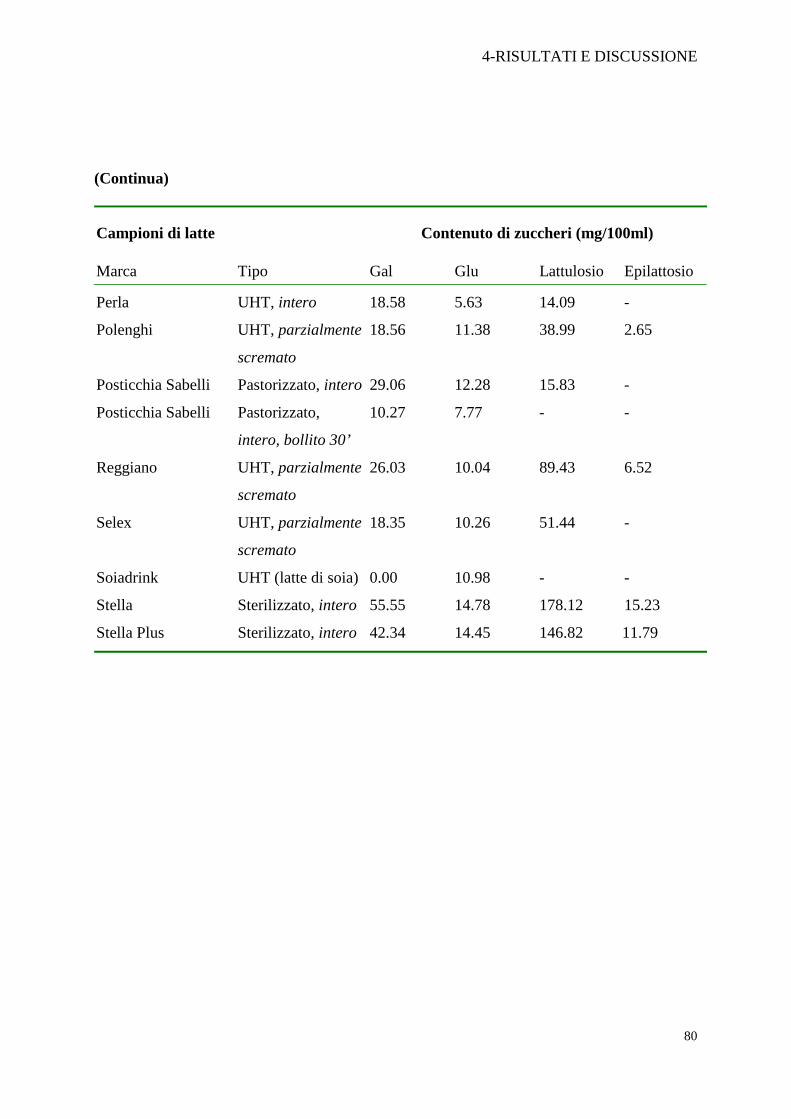
(Continua)
Campioni di latte Contenuto di zuccheri (mg/100ml)
Stella Plus Sterilizzato, intero 42.34 14.45 146.82 11.79
4-RISULTATI E DISCUSSIONE
81
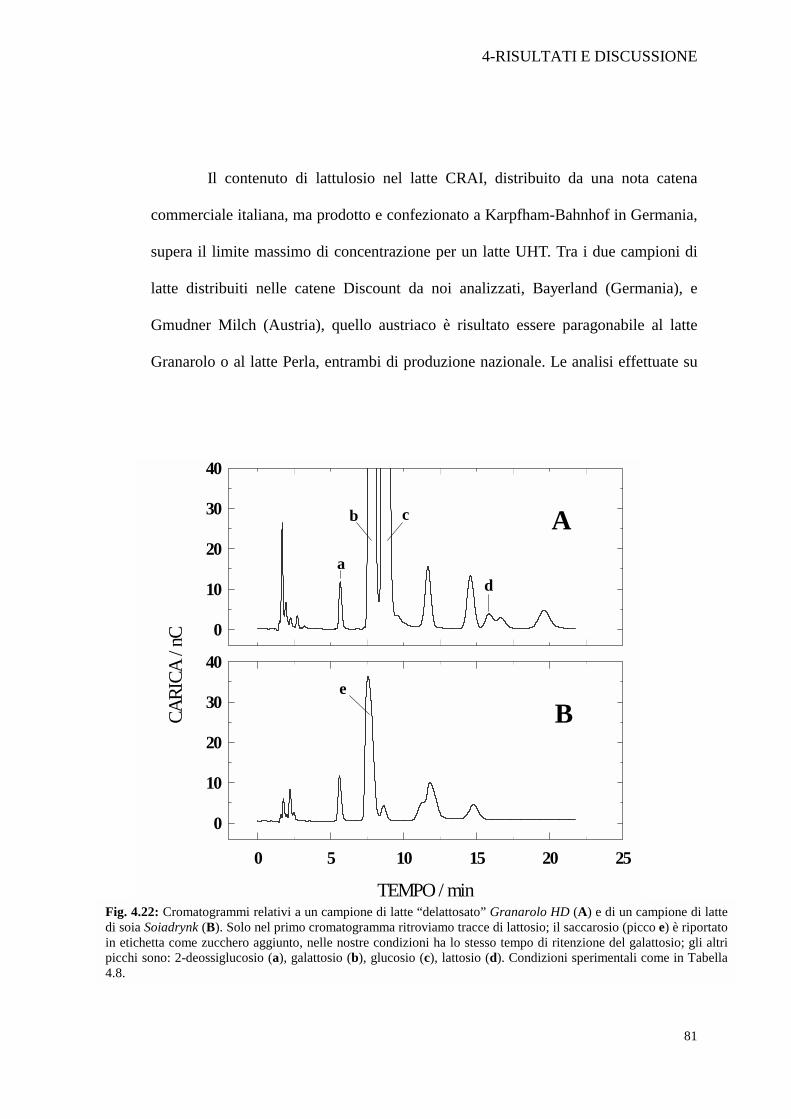
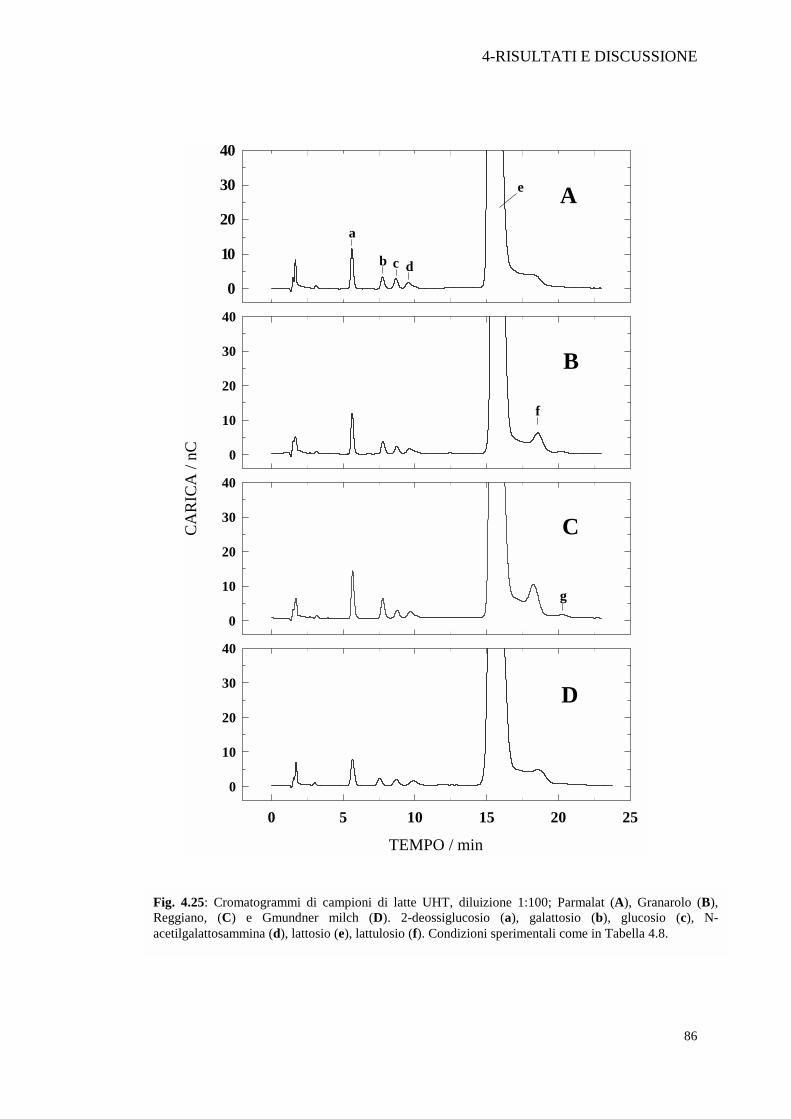
Il contenuto di lattulosio nel latte CRAI, distribuito da una nota catena
commerciale italiana, ma prodotto e confezionato a Karpfham-Bahnhof in Germania,
supera il limite massimo di concentrazione per un latte UHT. Tra i due campioni di
latte distribuiti nelle catene Discount da noi analizzati, Bayerland (Germania), e
Gmudner Milch (Austria), quello austriaco è risultato essere paragonabile al latte
Granarolo o al latte Perla, entrambi di produzione nazionale. Le analisi effettuate su
0
10
20
30
40
A
TEMPO / min
0 5 10 15 20 25
CARI
CA /
nC
0
10
20
30
40
B
a
b c
d
e
Fig. 4.22: Cromatogrammi relativi a un campione di latte “delattosato” Granarolo HD (A) e di un campione di latte di soia Soiadrynk (B). Solo nel primo cromatogramma ritroviamo tracce di lattosio; il saccarosio (picco e) è riportato in etichetta come zucchero aggiunto, nelle nostre condizioni ha lo stesso tempo di ritenzione del galattosio; gli altri picchi sono: 2-deossiglucosio (a), galattosio (b), glucosio (c), lattosio (d). Condizioni sperimentali come in Tabella 4.8.
4-RISULTATI E DISCUSSIONE
82
un latte “delattosato” (Granarolo HD) e su un latte di soia (Soiadrink), entrambi
destinati all’alimentazione di soggetti intolleranti al lattosio, hanno confermato
l’assenza di questo disaccaride nel latte di soia e la presenza in piccole tracce nel
latte “delattosato” (Figura 4.22 A).
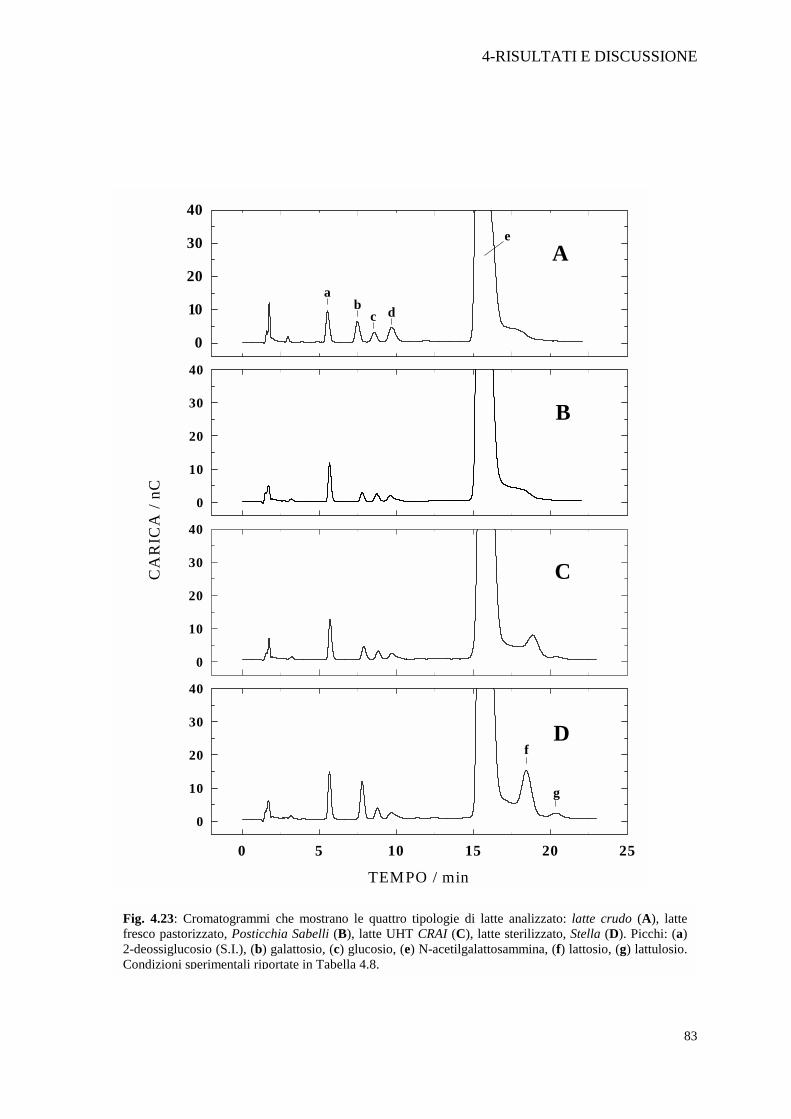
Nella Figura 4.23 A è rappresentato il cromatogramma di un campione di
latte crudo; sono presenti il galattosio, glucosio, N-acetilgalattosammina e lattosio.
Sono assenti sia il lattulosio che l’epilattosio. Le quantità relativamente elevate dei
glucidi presenti, potrebbero essere legate all’individualità del soggetto da cui è stato
prelevato il campione di latte. Non è stato infatti possibile prelevare il campione
dalla cisterna di raccolta del latte crudo, bensì direttamente dalla mungitrice
meccanica del singolo animale. La figura 4.23 B, relativa a un campione di latte
fresco pastorizzato, rivela un profilo di concentrazioni relativamente analogo al latte
crudo, ad indicare il minimo impatto che provoca il trattamento termico sulle
caratteristiche chimico fisiche del latte. Le figure 4.23 C e D mostrano, invece, i
cromatogrammi rispettivamente di un latte UHT e di un latte sterilizzato. In questi
due campioni è chiara la formazione di lattulosio ed epilattosio. Nel latte sterilizzato
è presente inoltre il galattosio in quantità relativamente più elevate di quelle degli
altri campioni. Tale differenza è probabilmente attribuibile ad una più spinta idrolisi
del lattosio rispetto al trattamento termico UHT.
4-RISULTATI E DISCUSSIONE
83
0
10
20
30
40
A
CA
RIC
A /
nC 0
10
20
30
40
0
10
20
30
40
TEMPO / min
0 5 10 15 20 25
0
10
20
30
40
B
C
D
ab
c d
e
f
g
Fig. 4.23: Cromatogrammi che mostrano le quattro tipologie di latte analizzato: latte crudo (A), lattefresco pastorizzato, Posticchia Sabelli (B), latte UHT CRAI (C), latte sterilizzato, Stella (D). Picchi: (a)2-deossiglucosio (S.I.), (b) galattosio, (c) glucosio, (e) N-acetilgalattosammina, (f) lattosio, (g) lattulosio.Condizioni sperimentali riportate in Tabella 4.8.
4-RISULTATI E DISCUSSIONE
84
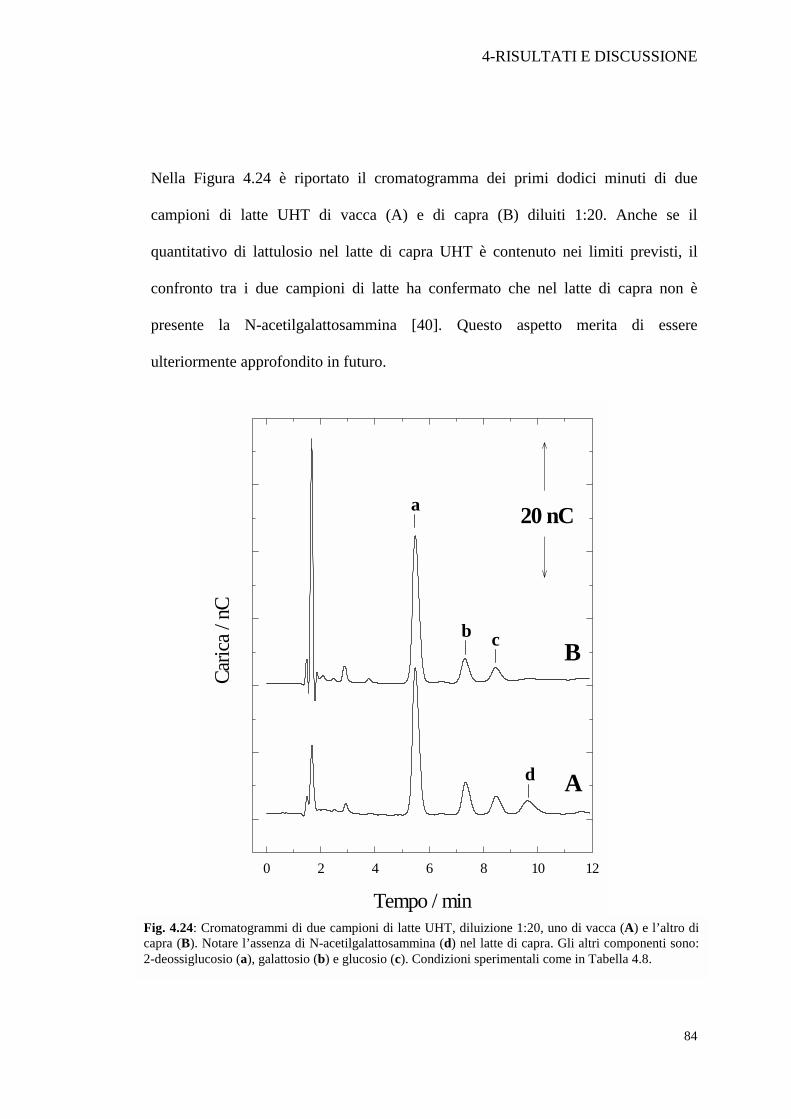
Nella Figura 4.24 è riportato il cromatogramma dei primi dodici minuti di due
campioni di latte UHT di vacca (A) e di capra (B) diluiti 1:20. Anche se il
quantitativo di lattulosio nel latte di capra UHT è contenuto nei limiti previsti, il
confronto tra i due campioni di latte ha confermato che nel latte di capra non è
presente la N-acetilgalattosammina [40]. Questo aspetto merita di essere
ulteriormente approfondito in futuro.
Tempo / min
0 2 4 6 8 10 12
Caric
a / n
C
a
b c
d A
B
20 nC
Fig. 4.24: Cromatogrammi di due campioni di latte UHT, diluizione 1:20, uno di vacca (A) e l’altro di capra (B). Notare l’assenza di N-acetilgalattosammina (d) nel latte di capra. Gli altri componenti sono: 2-deossiglucosio (a), galattosio (b) e glucosio (c). Condizioni sperimentali come in Tabella 4.8.
4-RISULTATI E DISCUSSIONE
85
Il picco riscontrato sul fronte del solvente è attribuibile molto probabilmente al mio-
inositolo, anch’esso presente in quantità maggiori nel latte di capra. Nelle nostre
condizioni cromatografiche però, sono più di uno gli alditoli che eluiscono con un
tempo di ritenzione prossimo a quello del tempo morto, di conseguenza
l’attribuzione del picco al mio-inositolo, presente tra l’altro in tutti i campioni
analizzati, non può essere certa.
Un’ultima considerazione può essere fatta, relativamente al trattamento
termico, sulla qualità del latte commercializzato da grandi marchi commerciali e da
catene discount. Non è così immediato infatti il binomio discount = bassa qualità,
anzi in alcuni casi la quantità di lattulosio presente nel latte “discount” è di gran
lunga inferiore a quella riscontrata in altre marche di fascia media commercializzate
nelle catene di distribuzione quali Conad, Coop o Crai. È interessante aver stabilito
per quest’ultima in particolare, che la produzione del latte venduto con il marchio
Crai è affidata allo stabilimento Rottaler Milchwerk e G., Karpfham-Bahnhof,
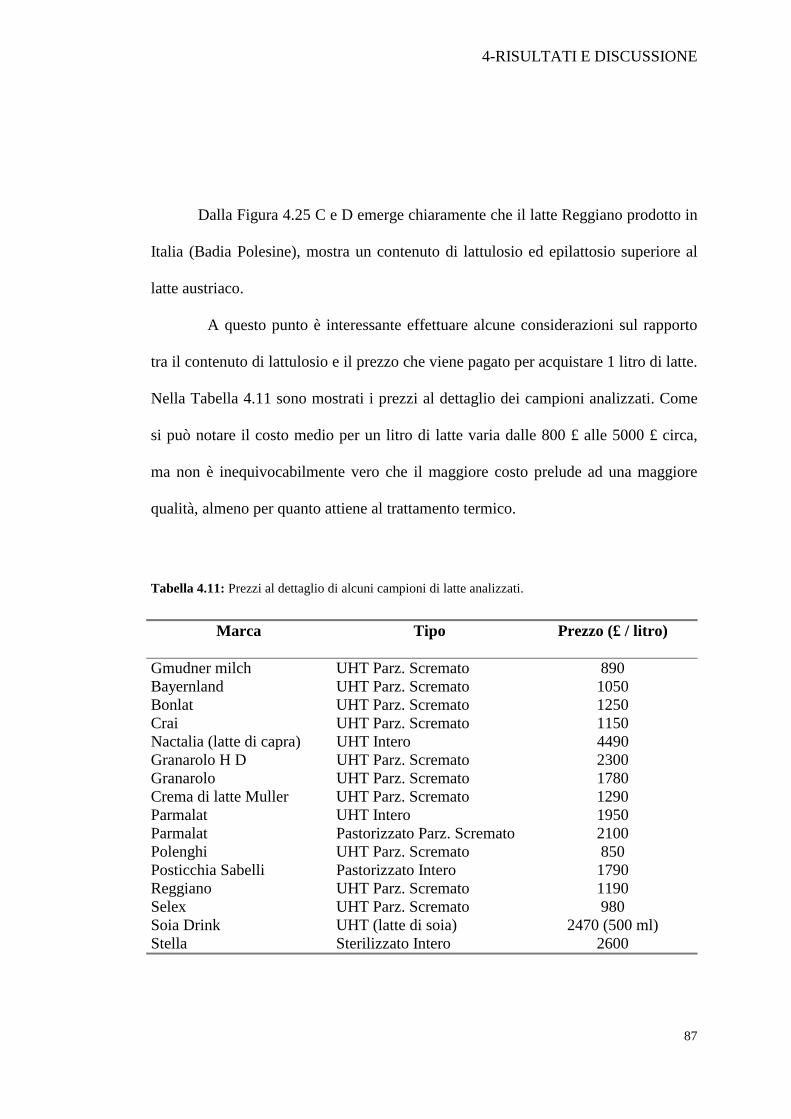
Bavaria. Il costo per un litro di latte Crai supera di poco quello di un litro di latte
discount, ma la quantità di lattulosio presente nel latte “più caro” è maggiore di
quella presente nel secondo. Nella Figura 4.25 vengono posti a confronto un latte che
si pone nella fascia di prezzo medio alta, Parmalat UHT intero (A); latte di costo
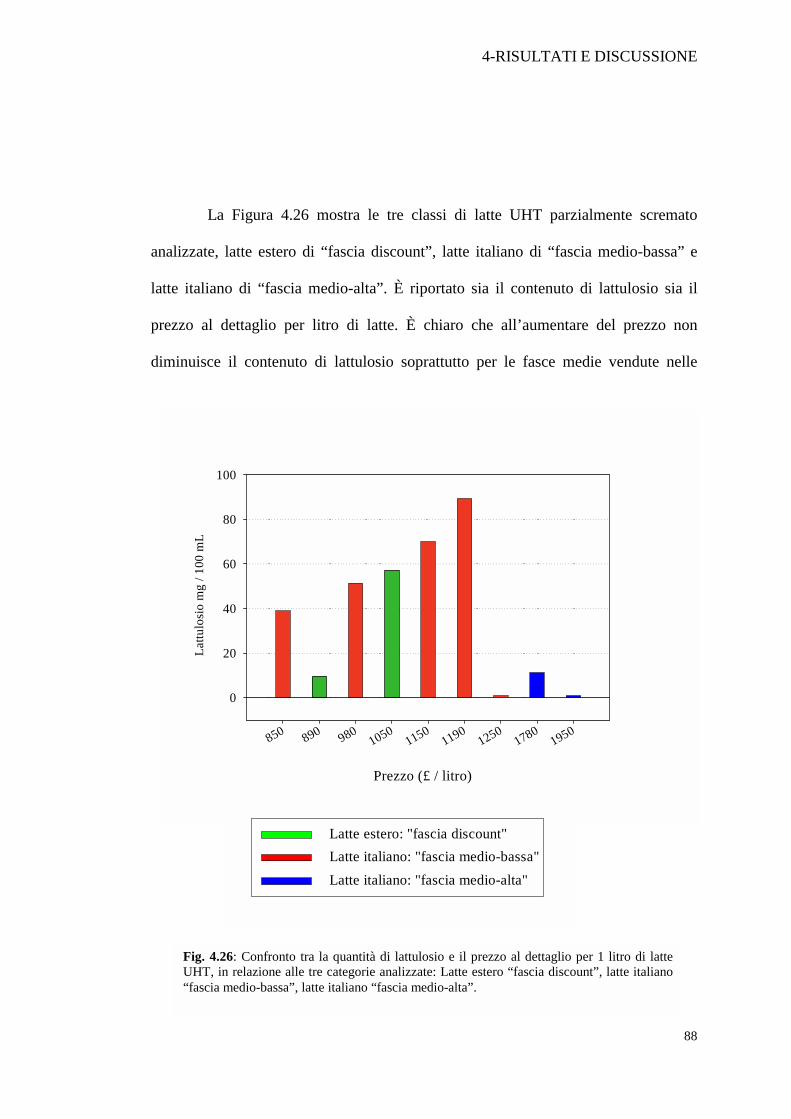
Fig. 4.26: Confronto tra la quantità di lattulosio e il prezzo al dettaglio per 1 litro di latteUHT, in relazione alle tre categorie analizzate: Latte estero “fascia discount”, latte italiano“fascia medio-bassa”, latte italiano “fascia medio-alta”.
4-RISULTATI E DISCUSSIONE
89
grandi distribuzioni che sono quindi le dirette concorrenti del latte di qualità
discount; così il campione di latte Bonlat che costa £ 1250/litro è un esempio
dell'incidenza del marchio sulla determinazione del prezzo finale. Dall’etichetta
infatti risulta prodotto nello stabilimento Parmalat di Collecchio (Pr), però ha un
costo di circa 500 £ in meno del latte Parmalat £ 1950/litro. I motivi per cui il latte
estero riesce a spuntare un prezzo minore anche a fronte dell’elevata incidenza del
costo per il trasporto, sono probabilmente da attribuire agli accordi comunitari per il
sostegno alle produzioni di ogni paese. Questo fatto tende a favorire l'importazione
di latte estero, a danno evidentemente del mercato interno.
Come già evidenziato da Andrews [41], infine, le prove effettuate su
campioni di latte della stessa ditta produttrice, in tempi diversi e su campioni di latte
con diverso contenuto in grasso, non hanno mostrato differenze significative nel
contenuto dei glucidi analizzati mediante HPAEC-PAD.
4-RISULTATI E DISCUSSIONE
90
4.10. CONCLUSIONI
Nel presente lavoro di tesi è stata messa a punto una nuova metodica per la
determinazione del lattulosio nel latte trattato termicamente, mediante cromatografia
a scambio anionico con rivelazione amperometrica pulsata. La scelta delle condizioni
cromatografiche è stata effettuata con lo scopo di ottenere una efficiente e rapida
separazione di tutti i principali glucidi presenti nel campione.
La scelta di utilizzare una colonna a scambio anionico ad alto grado di
reticolazione, quale la CarboPac PA10, ha permesso di migliorare la risoluzione tra
lattulosio e lattosio presenti nel latte in concentrazioni 1:1000. Una buona
separazione è stata anche ottenuta con una colonna CarboPac PA1. L’aggiunta di
bario acetato alla soluzione alcalina, ci ha permesso di utilizzare una fase mobile
diluita di NaOH (10 mM) con la quale è possibile lavorare per più giorni senza dover
rigenerare la colonna dopo ogni corsa cromatografica. Questo perché il bario induce
la precipitazione del carbonato presente in fase mobile sotto forma di sale insolubile.
Induce inoltre un aumento della risposta amperometrica, molto utile per il buon
funzionamento del rivelatore elettrochimico pulsato. L’aggiunta del sale sotto forma
di acetato ha infine garantito una riduzione dei tempi di analisi con una durata per
ogni corsa cromatografica di soli 25 minuti.
Interessanti considerazioni possono essere dedotte dall’analisi dei campioni
di latte. È infatti stato dimostrato che a fronte di un minor costo del latte “discount”,
non sempre corrisponde una bassa qualità del prodotto. Si è bensì verificato il
contrario. Dal confronto tra i campioni di latte “nazionali” e i campioni di latte
4-RISULTATI E DISCUSSIONE
91
prodotti per conto di ditte italiane all’estero, risulta che il livello di lattulosio è più
elevato per il latte "estero". Questo suggerisce che tale prodotto è sottoposto ad un
trattamento termico più drastico o eventualmente addizionato con latte in polvere.
La possibilità di espandere la metodica sviluppata per la determinazione di tutta una
serie di glucidi e loro derivati presenti nel latte, possibili indicatori di altri trattamenti
o difetti della materia prima, è attualmente oggetto di studio nel nostro laboratorio.
RINGRAZIAMENTI
Porgo un sentito ringraziamento al Prof. Tommaso Cataldi per l’assidua
presenza e disponibilità mostrata durante tutto il lavoro di tesi e al Dipartimento di
Produzione Vegetale per avermi ospitato e per aver consentito l’utilizzazione del
cromatografo Dionex.
Un ultimo ringraziamento alla Dott.ssa Cristiana Campa, preziosa e attenta
assistente in molte prove in laboratorio.
BIBLIOGRAFIA
4-RISULTATI E DISCUSSIONE
92
1 Charles Alais, Scienza del latte – Principi di tecnologia del latte e dei derivati. Ed.