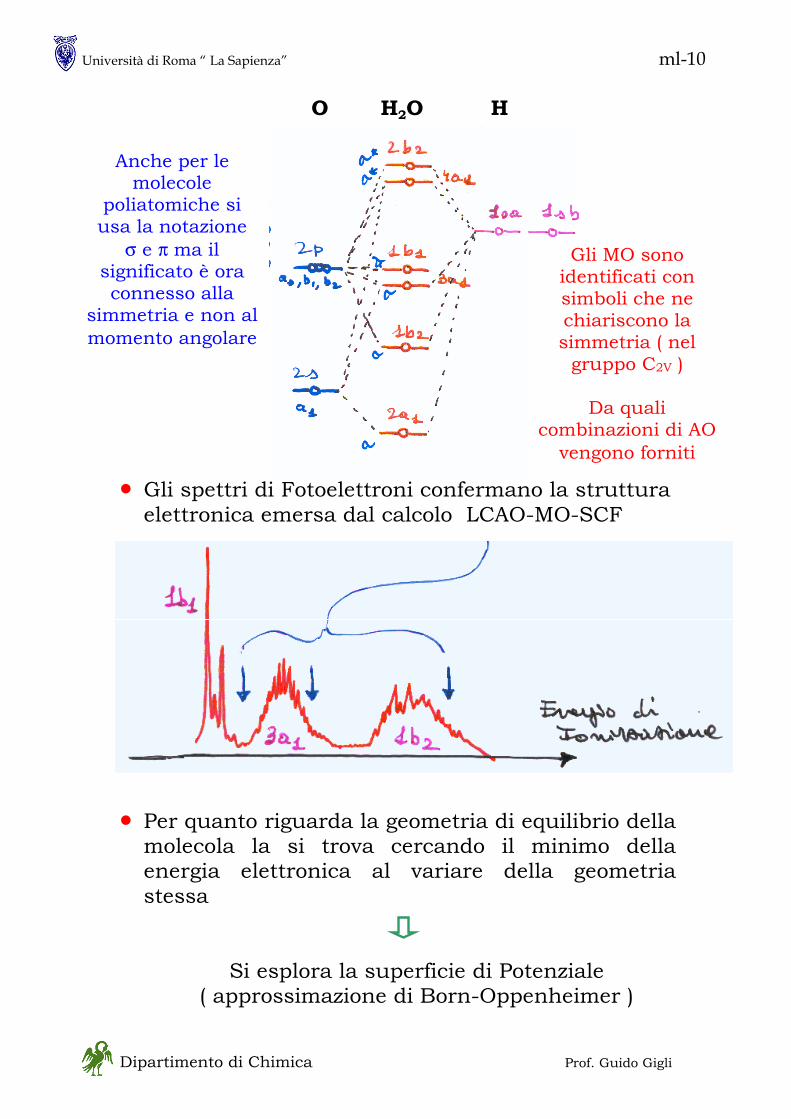
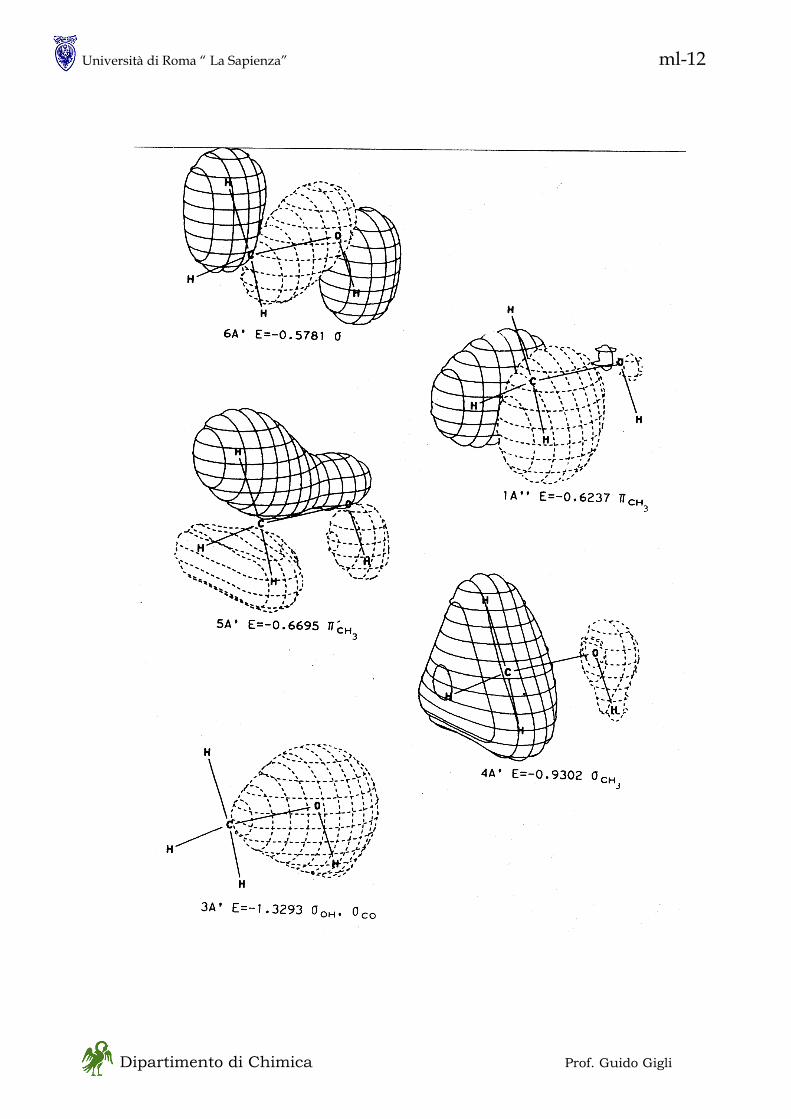
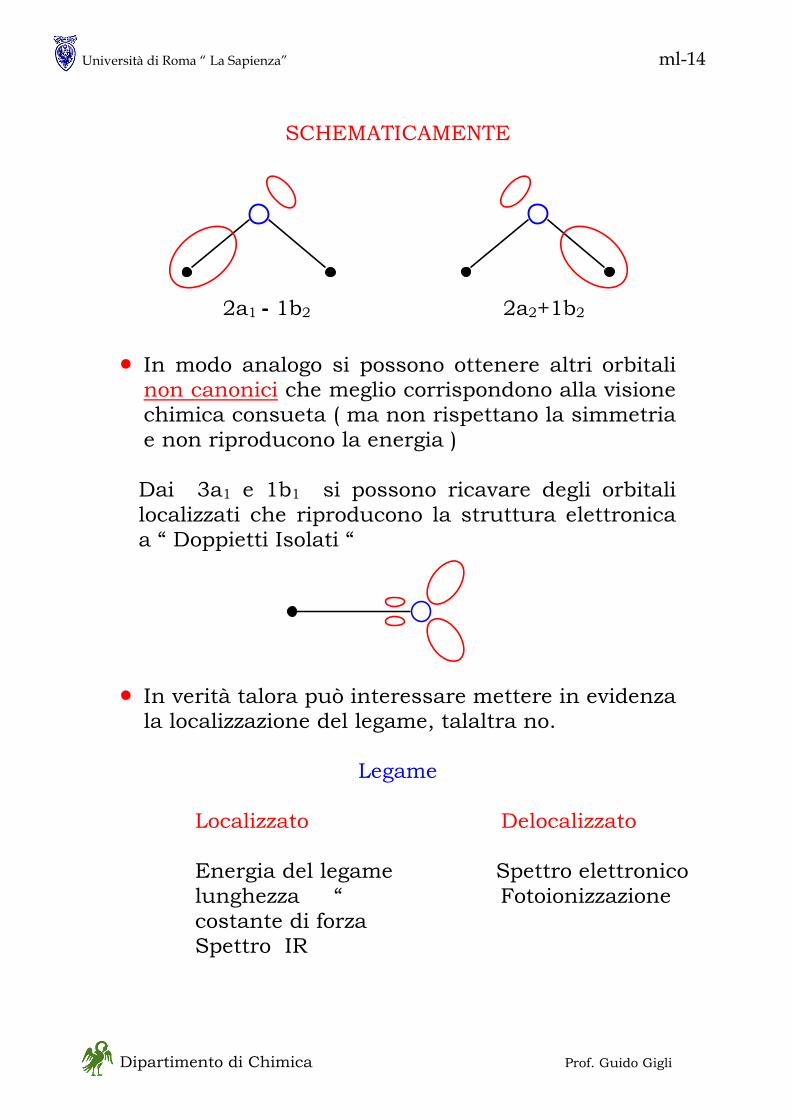
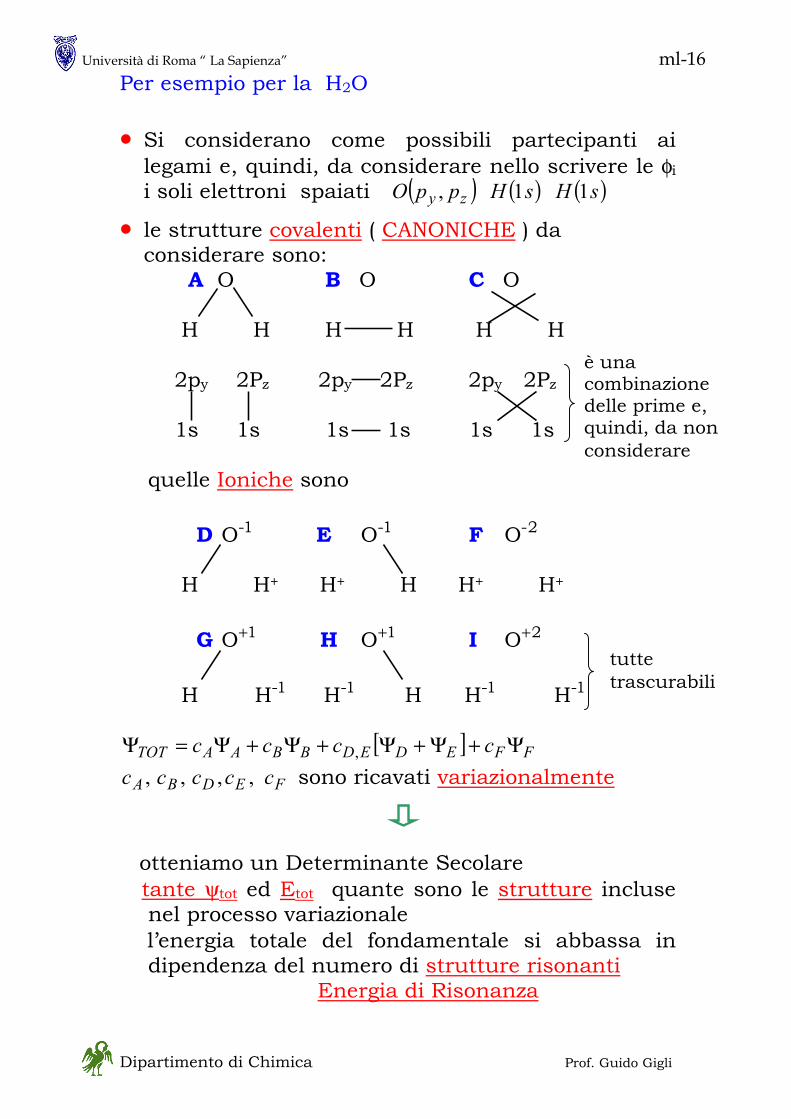
in - 1 Dipartimento di Chimica Prof. Guido Gigli MECCANICA QUANTISTICA • Ne esistono, sostanzialmente, due formulazioni, entrambe sviluppate fra il 1925 ed il 1930 ed usate in particolare modo per i sistemi chimici - Meccanica Matriciale :essenzialmente dovuta ad Heisenberg è basata sulla asssociazione fra osservabili fisiche e matrici - Meccanica Ondulatoria :essenzialmente dovuta a Schroedinger è basata sulla associazione di ogni particella con una “funzione d’onda” • Si tratta di formulazioni equivalenti unificate da Dirac (1930) in una forma assai simile alla meccanica matriciale La trattazione di Dirac è più generale e più potente della meccanica ondulatoria ma è più astratta • Non seguiremo lo sviluppo storico della meccanica quantistica perché la pratica attuale se ne discosta alquanto; tuttavia vi sono alcuni concetti chiave che sono meglio comprensibili in una prospettiva storica. Tra questi la natura DUALISTICA dei sistemi fisici ONDA – PARTICELLA (materiale)
Transcript
in - 1
Dipartimento di Chimica Prof. Guido Gigli
MECCANICA QUANTISTICA • Ne esistono, sostanzialmente, due formulazioni,
entrambe sviluppate fra il 1925 ed il 1930 ed usate in particolare modo per i sistemi chimici
- Meccanica Matriciale :essenzialmente dovuta ad
Heisenberg è basata sulla asssociazione fra osservabili fisiche e matrici
- Meccanica Ondulatoria :essenzialmente dovuta
a Schroedinger è basata sulla associazione di ogni particella con una “funzione d’onda”
• Si tratta di formulazioni equivalenti unificate da
Dirac (1930) in una forma assai simile alla meccanica matriciale
La trattazione di Dirac è più generale e più potente della meccanica ondulatoria ma è più astratta • Non seguiremo lo sviluppo storico della meccanica
quantistica perché la pratica attuale se ne discosta alquanto; tuttavia vi sono alcuni concetti chiave che sono meglio comprensibili in una prospettiva storica. Tra questi la natura
DUALISTICA dei sistemi fisici
ONDA – PARTICELLA (materiale)
in - 2
Dipartimento di Chimica Prof. Guido Gigli
Particelle ed onde Spettro di emissione del corpo nero • Ogni corpo ad una determinata temperatura
emette luce ( radiazione) con una intensità che varia con la frequenza (distribuzione delle frequenze)
• Questa distribuzione dipende dalla natura del materiale e della sua superficie ma ne è indipendente al limite ideale di
CORPO NERO
un corpo nero assorbe piu’ di qualsiasi superficie un corpo nero emette piu’ di qualsiasi superficie
radiatore integrale
• si chiama corpo nero perché un raggio luminoso che vi entri viene assorbito e riemesso tante volte (viene riflesso) e tutta la sua energia viene integralmente convertita in agitazione termica del materiale
• la luce originariamente entrata non “esiste” piu’
• la cavità raggiunge l’equilibrio termodinamico
• la luce che esce dal corpo nero dipende in intensita’ e frequenza unicamente dalla sua temperatura
in - 3
Dipartimento di Chimica Prof. Guido Gigli
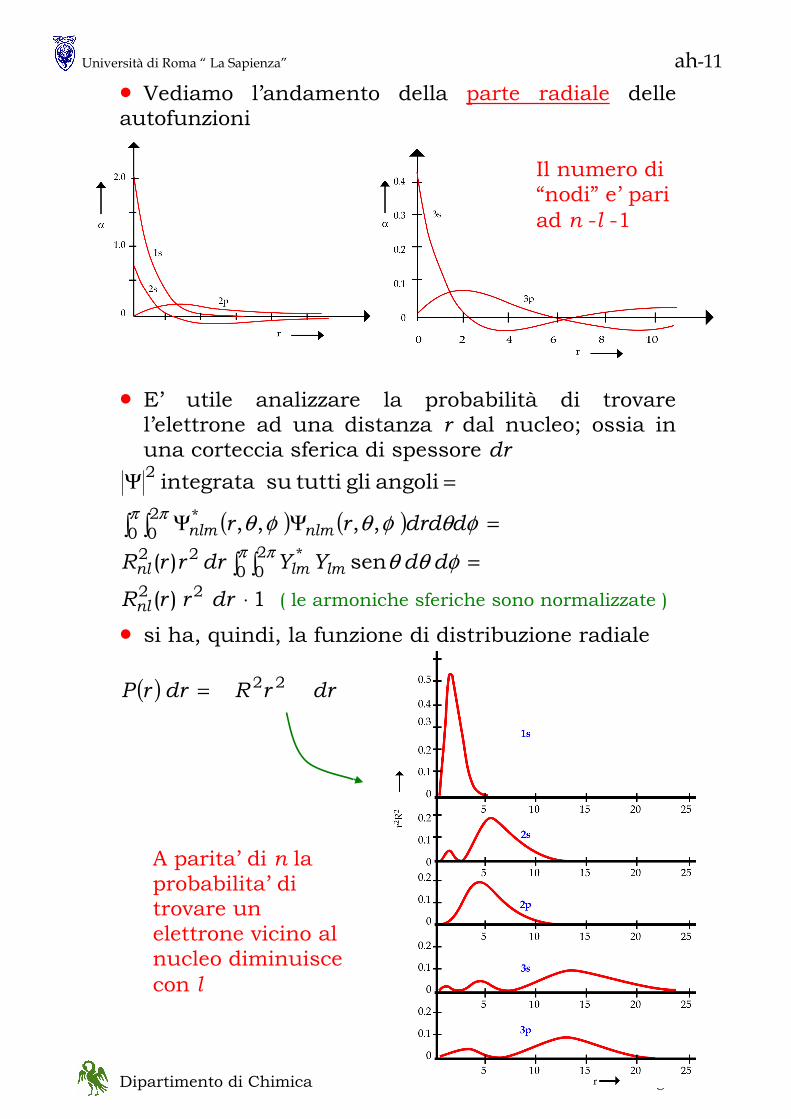
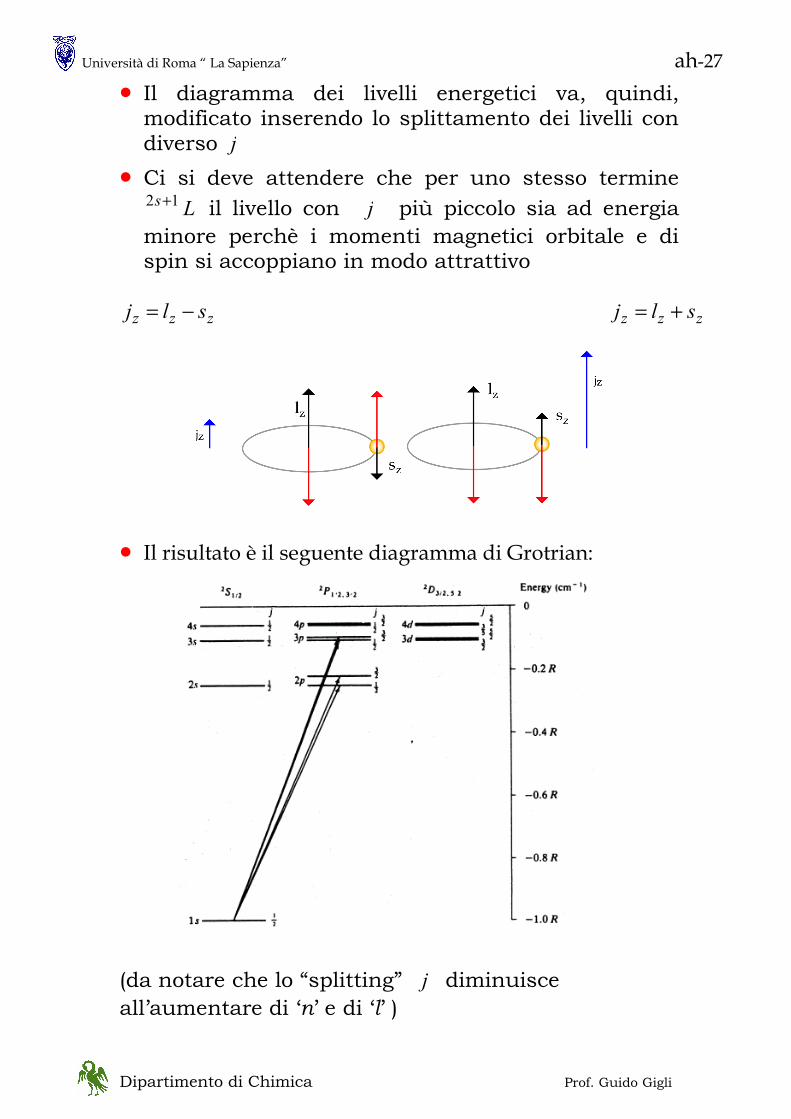
• lo spettro della radiazione emessa da un corpo nero ha questo aspetto
N.B. lo spostamento in νmax (λmax) con la temperatura va sotto il nome di legge di Wien ( Nobel 1911): λmax T = cost
Questo complesso di osservazioni non avevano spiegazione con le teorie classiche (Raleigh) che prevedevano un aumento senza limiti della intensita’ della luce emessa all’aumentare della frequenza della stessa: “catastrofe ultravioletta” • Nel 1900 Planck riuscì a spiegare l’andamento
sperimentale di ( )λfI = ammettendo, come Raleigh, che gli atomi del materiale della cavità oscillino e che sia la loro oscillazione a rendere possibile l’assorbimento e la emissione di radiazione (si comportano come gli elettroni nelle antenne)
MA postulando anche che queste oscillazioni non possano avvenire per qualsiasi energia ma soltanto per multipli di unità discrete
Quanti ( dal latino quantum )
sJhhE ⋅≅=Δ −34106.6ν
in - 4
Dipartimento di Chimica Prof. Guido Gigli
• se la distribuzione di Boltzmann si può applicare al numero di questi “quanti” l’aumento catastrofico di intensità all’aumentare di ν non ha più luogo
• La ragione qualitativa di ciò è che ad alte energie un quanto “contiene” tanta energia da rendere altamente improbabile la sua esistenza
( )( )kToioi enn /εε −−=
viceversa nella teoria classica la energia dipende dalla ampiezza di oscillazione (non dalla frequenza)
L’effetto Fotoelettrico • Emissione di elettroni da un metallo illuminato
1. Emissione istantanea 2. E cinetica dei fotoelettroni dipendente da ν
3. ν minima di fotoemissione • Le osservazioni sperimentali non trovavano
spiegazioni con le teorie classiche
in - 5
Dipartimento di Chimica Prof. Guido Gigli
INFATTI (secondo queste ultime)
1. ci deve essere un certo ritardo, cioè un intervallo di tempo, durante il quale gli elettroni oscillando in sincrono con la perturbazione (la radiazione) assorbono man mano energia aumentando la ampiezza di oscillazione fino ad averne abbastanza da sfuggire
2. – 3. - secondo l’esperimento la energia è concentrata in
piccole zone anziché essere distribuita su tutto il fronte d’onda; viceversa secondo Maxwell la energia dipende dall’ampiezza dell’onda e non dalla sua frequenza
• Einstein (1905) postulò che la radiazione elettromagnetica consista di un fascio di particelle, FOTONI, tutte con la medesima velocità
18100.3 −≅ msc Ognuno di questi fotoni ha
una frequenza caratteristica ν
una energia ωωπ
ν h===2hhE
sJ ⋅= −341005.1h
Il fotone, con velocità c , deve seguire la teoria della relatività ed il suo momento p risulta:
( )21
4222 cmcpE +=
cch
cEp ων h
===
Introducendo il numero d’onda c
k ω=
kp h= l’esistenza di questo momento è confermata dall’effetto COMPTON (1924)
0
in - 6
Dipartimento di Chimica Prof. Guido Gigli
• in definitiva
221 mvhE fotone +Φ== ν
work function (minima energia necessaria
per uscire dal metallo) poichè gli elettroni in un metallo hanno molte energie diverse alcuni necessitano di maggiore energia di altri ed ½ mv2 è la massima energia cinetica degli elettroni fotoemessi La natura Ondulatoria delle particelle • De Broglie (1924),simmetricamente a quanto fatto
da Einstein per la luce, suggerì di associare un comportamento ondulatorio al moto di una particella. Egli postulò che ad una particella con momento lineare p sia associata una onda di lunghezza d’onda λ ed energia E
ph
k==
πλ 2 ωh=E kp h=
• La natura ondulatoria del moto di particelle è stata confermata sperimentalmente per
• Vediamo ora un esperimento che illustra la natura
dualistica onda – corpuscolo sia della luce che delle particelle materiali (per esempio gli elettroni)
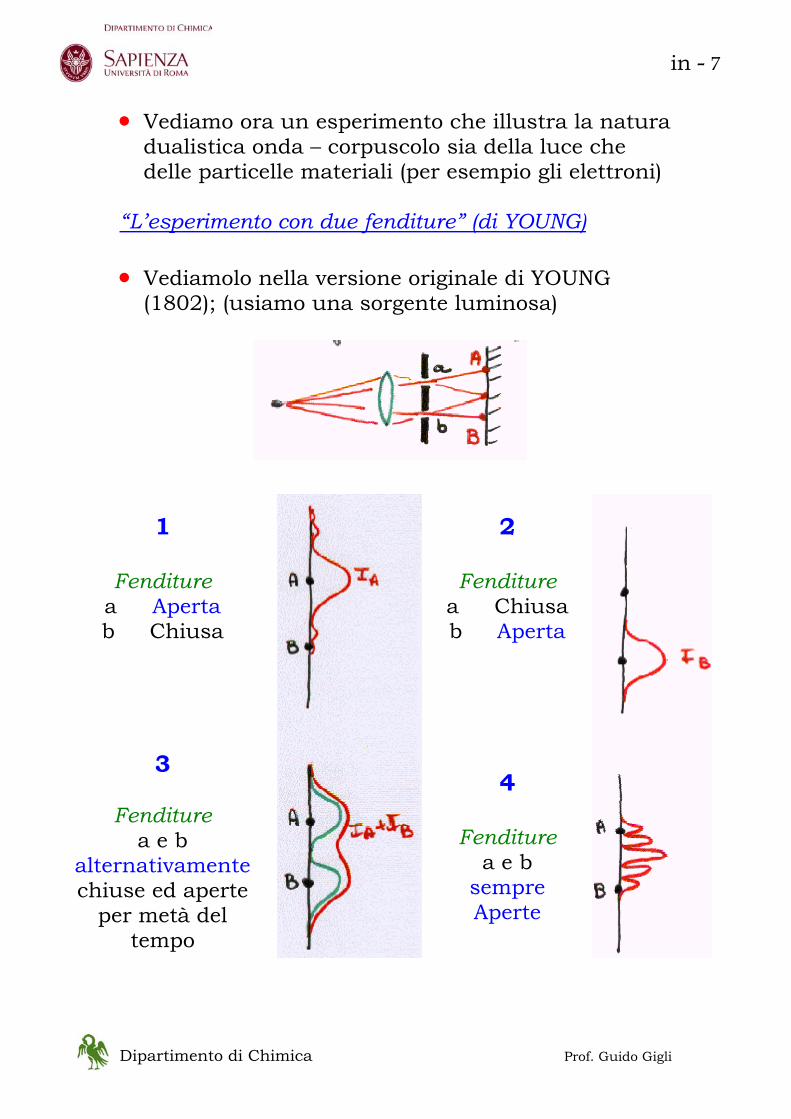
“L’esperimento con due fenditure” (di YOUNG) • Vediamolo nella versione originale di YOUNG
(1802); (usiamo una sorgente luminosa)
1
Fenditure
a Aperta b Chiusa
3
Fenditure a e b
alternativamente chiuse ed aperte
per metà del tempo
2
Fenditure
a Chiusa b Aperta
4
Fenditure a e b
sempre Aperte
in - 8
Dipartimento di Chimica Prof. Guido Gigli
• Queste osservazioni sono spiegabili con la
interpretazione ondulatoria della luce e le sue proprietà di interferenza
MA
l’interferenza non si spiega con la luce costituita da fotoni
INFATTI
Se la luce è costituita da fotoni è ragionevole pensare che ogni fotone passi per la fenditura “a” oppure per quella “b”
Si può confermare questa ipotesi con una sorgente molto debole (tale che un solo fotone per volta venga emesso) e due rivelatori vicino e dietro le fenditure “a” e “b”
I rivelatori registrano un fotone per “a” oppure per “b” e mai per entrambi simultaneamente
Metà dei fotoni passano da “a” e metà da “b”
Tutto ciò è coerente con la interpretazione corpuscolare
• Ci si domanda:
Come si possono influenzare vicendevolmente i fotoni che passano per fenditure diverse?
in - 9
Dipartimento di Chimica Prof. Guido Gigli
• Ripetiamo l’esperimento con
fenditure a e b aperte un fotone per volta emesso dalla Sorgente con
frequenza tanto piccola (periodo tanto lungo) da escludere che i fotoni possano influenzarsi l’un l’altro
Si osserva la solita figura di interferenza
• Usiamo ora uno schermo in grado di rivelare la posizione dei punti di arrivo dei fotoni Si osserva:
1. Ogni fotone colpisce lo schermo in un unico punto
2. La figura di interferenza si forma come conseguenza dell’accumularsi dei vari singoli impatti dei fotoni
Il comportamento di ogni singolo fotone non è prevedibile
La frequenza (densità) degli impatti in ogni punto dello schermo fornisce le bande di interferenza
CIOE’ La figura di interferenza ci fornisce la distribuzione di probabilità delle posizioni dei punti di arrivo dei fotoni
L’interferenza persiste con qualsiasi intensità di
luce
in - 10
Dipartimento di Chimica Prof. Guido Gigli
• nel caso dell’esperimento 3 i fotoni che passano uno alla volta per “a” o per “b” danno origine, in modo statistico, alle figure IA e IB e, quindi, all’effetto complessivo IA + IB
MA
• Se lasciamo a e b aperti con un rivelatore in a che ci “dice” se ciacun fotone è passato da a o da b caso 5
NON si osserva la figura di interferenza ma la semplice IA + IB
l’atto di accertare da quale fenditura passa ogni fotone ha il medesimo effetto del chiudere l’altra fenditura
IN SINTESI • Se un fotone passa indisturbato attraverso le
fenditure mostra un comportamento ondulatorio e si osserva una figura di interferenza ( o di diffrazione con una sola fenditura). Ogni fotone colpisce lo schermo in un punto specifico, in accordo con la natura corpuscolare, e la figura che risulta dall’arrivo di molti fotoni è una distribuzione di probabilità
• Se un fotone è costretto a (oppure è osservato) passare attraverso una specifica fenditura le figure di interferenza non vengono osservate ed il suo comportamento è più simile a quello di una particella
• Esperimenti analoghi sono stati fatti con elettroni anzichè fotoni
in - 11
Dipartimento di Chimica Prof. Guido Gigli
• Abbiamo visto una serie di osservazioni sperimentali non spiegabili compiutamente con la fisica classica
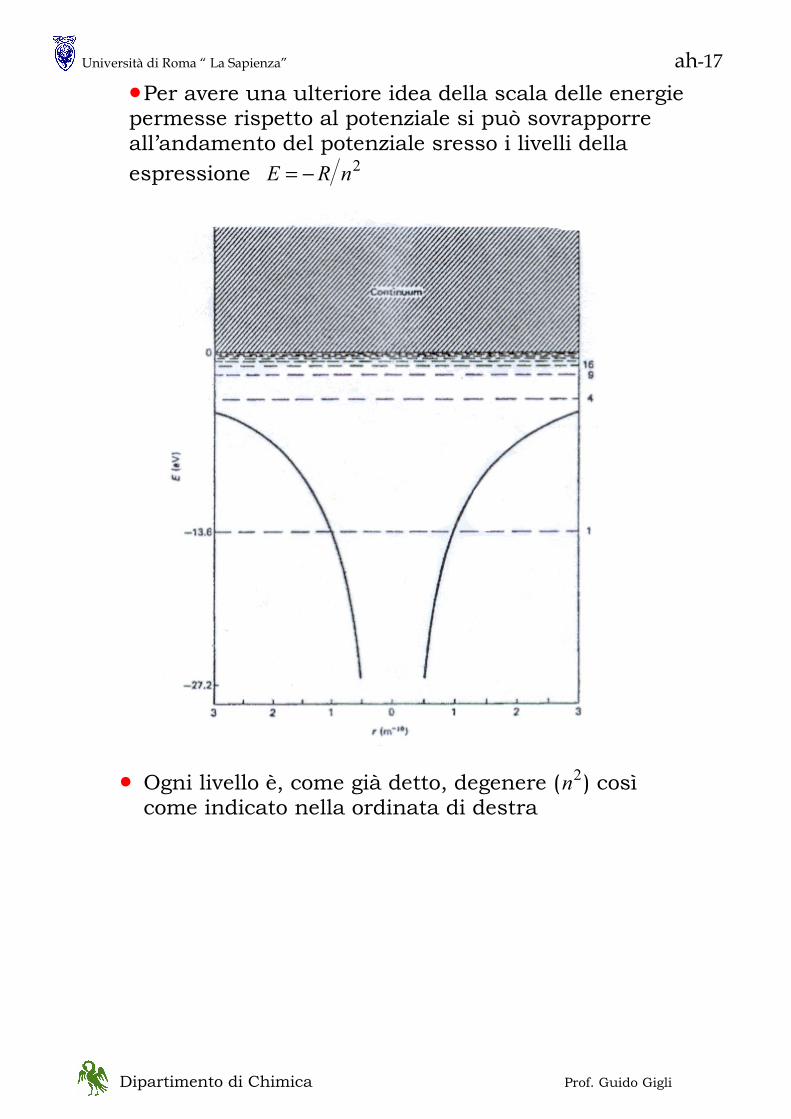
• Una spinta decisiva all’introduzione di teorie di tipo quantistico venne dall’osservazione che la luce viene emessa dagli atomi con uno spettro discreto
In particolare, per l’Idrogeno vale la semplice relazione:
⎟⎟⎠
⎞⎜⎜⎝
⎛−= 2
221
11nn
RHν
Più in generale vale il principio di Raleygh-Ritz
21 TT −=ν Suggerisce che i livelli energetici
degli atomi assumano unicamente valori discreti
Bohr ne diede una “spiegazione” imponendo al momento angolare degli elettroni di assumere valori discreti
• Vediamo ora un esperimento che mette in luce drammatici effetti quantistici ( e che è stato, anche, il primo esperimento di natura non ottica a mostrarli)
RH = 109700 cm-1
costante di Rydberg n1,n2 numeri interi
in - 12
Dipartimento di Chimica Prof. Guido Gigli
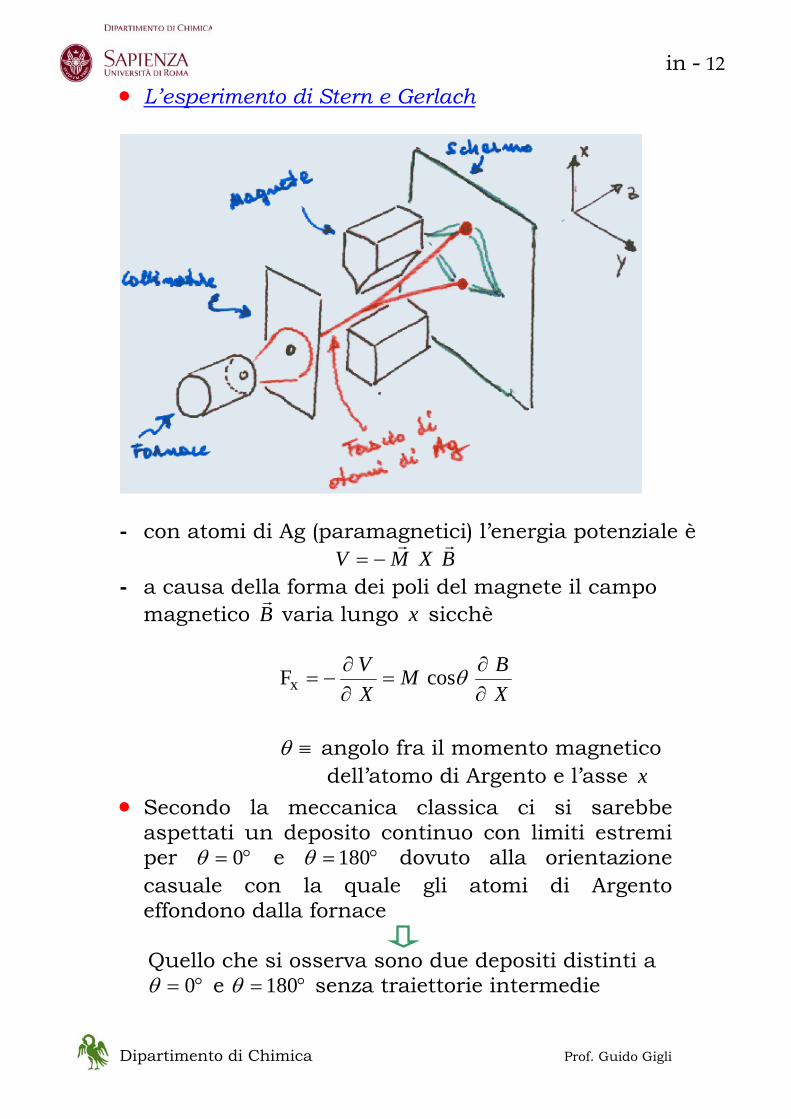
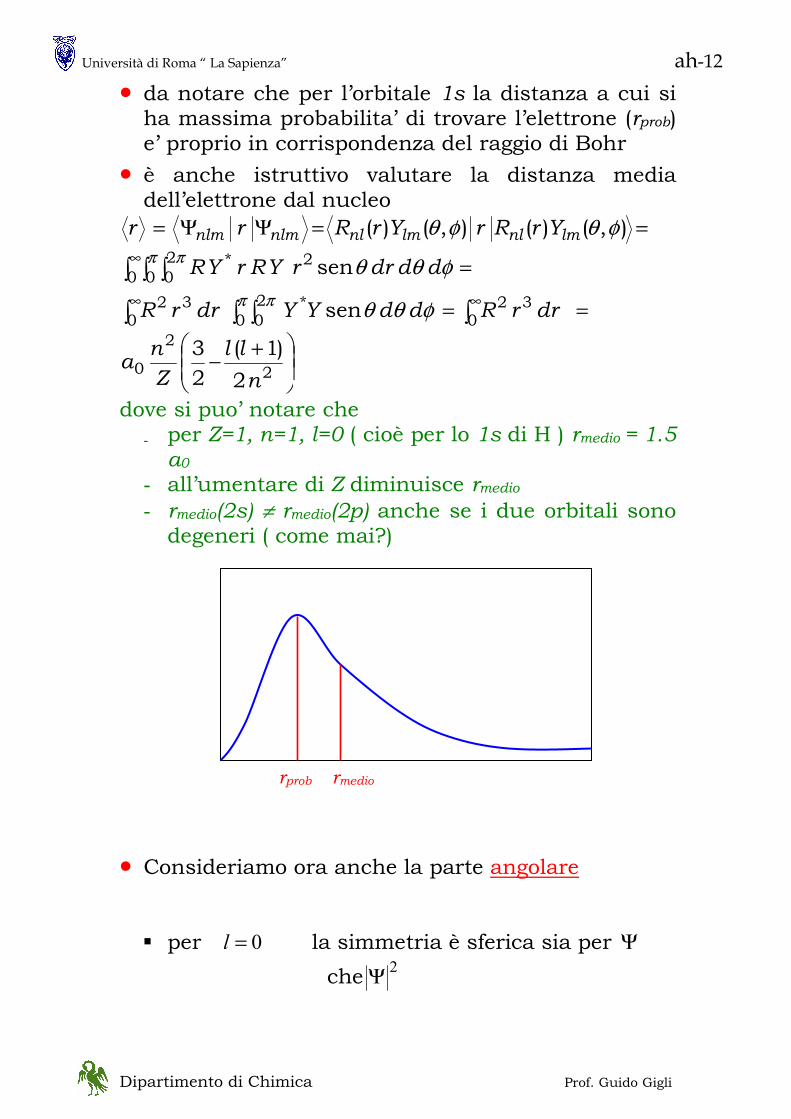
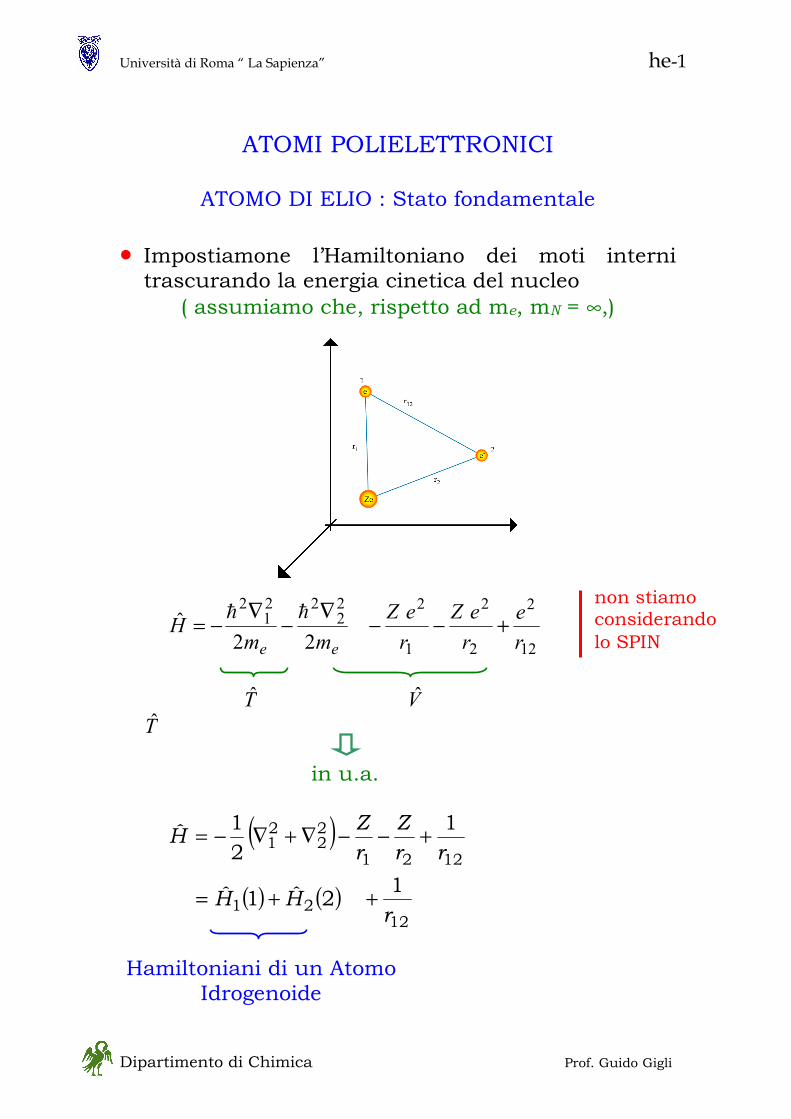
• L’esperimento di Stern e Gerlach
- con atomi di Ag (paramagnetici) l’energia potenziale è
BXMVrr
−= - a causa della forma dei poli del magnete il campo
magnetico Br varia lungo x sicchè
XBM
XV
∂∂
=∂∂
−= θcosFx
≡θ angolo fra il momento magnetico
dell’atomo di Argento e l’asse x • Secondo la meccanica classica ci si sarebbe
aspettati un deposito continuo con limiti estremi per °= 0θ e °= 180θ dovuto alla orientazione casuale con la quale gli atomi di Argento effondono dalla fornace
Quello che si osserva sono due depositi distinti a
°= 0θ e °= 180θ senza traiettorie intermedie
in - 13
Dipartimento di Chimica Prof. Guido Gigli
• questo esperimento mostra che: 1. Anche se i momenti magnetici degli atomi sono
orientati a caso nel momento della effusione essi sono “trovati” essere soltanto paralleli o antiparalleli
2. Le orientazioni possibili sono quantizzate, nel senso che soltanto alcuni valori sono osservati
3. L’atto della misura influenza il risultato della misura stessa (la direzione di quantizzazione è determinata dalla direzione del campo magnetico)
• Varianti di questo esperimento mettono in luce altri effetti di natura quantistica
SCHEMATICAMENTE
a) Un deposito E’ quello che ci si aspetta: il primo “Filtro” ha selezionato le orientazioni parallele
ad x ed il secondo non ha alcun effetto
b) Due depositi Anche qui accade cio’ che che ci si aspetta: il primo “Filtro” che agisce lungo x non ha
effetto su quanto fa il secondo che opera lungo y
c) Due depositi Non e’ quanto ci si aspetta: il primo magnete non ha operato come un filtro infatti le due
orientazioni lungo x sono ricomparse come conseguenza dell’aver agito con il magnete lungo y
in - 14
Dipartimento di Chimica Prof. Guido Gigli
• Il passaggio degli atomi attraverso il secondo magnete ha distrutto la informazione del primo magnete
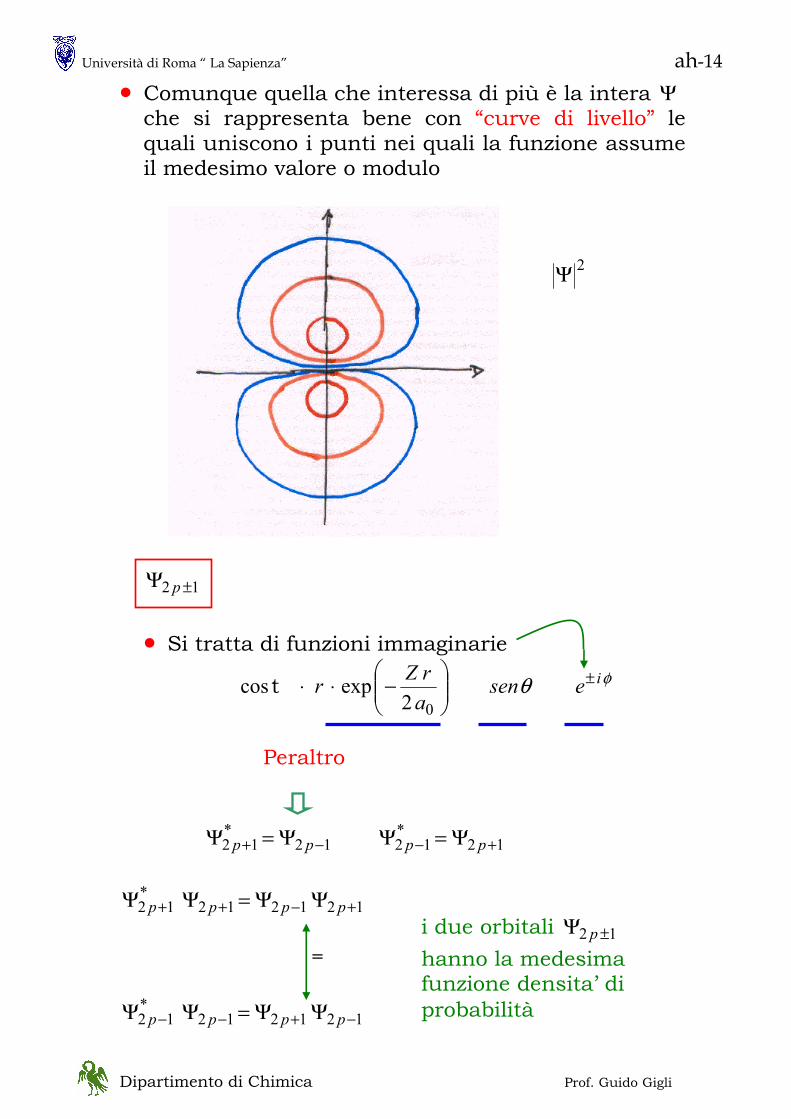
• Abbiamo visto che per interpretare pienamente la natura della materia è utile considerare anche la sua natura ondulatoria. Vediamo, allora, alcune caratteristiche del moto delle onde
MOTO DELLE ONDE
Onda Piana
( )λπ
λπ xxAx 2cos2cos ==Ψ
( ) ( )[ ] ( )[ ]txxxtx v−=−=Ψ λπ
λπ 2
02 coscos,
λvvelocità ==v
frequenza
( ) ( ) ⎟⎠⎞
⎜⎝⎛ −=−=Ψ txtvxtx ν
λπλ
λπ 2cos2cos,
λπ2
=k numero d’onda ( vettore d’onda )
πνω 2= velocità angolare
( ) ( )txktx ω−=Ψ cos, fase
kω
=fasev ( velocità di fase )
( ) costcost+=
=−tkx
txkωω
l’onda si muove verso x crescenti con velocita’ costante ω/k
in - 15
Dipartimento di Chimica Prof. Guido Gigli
• Abbiamo usato la funzione coseno ma si possono usare
( ) ( )[ ]txkitx ω+−=Ψ∗ exp, Onda Composta • E’ la sovrapposizione ( la somma ) di un certo
numero di onde piane
( ) ( )[ ]∑ −=Ψj
jjj txkiAtx ωexp,
dove ogni onda j ha la sua velocità di fase jjfase kω=v
• per esempio consideriamo una onda composta da due
( )[ ]txki 111 exp ω−=Ψ ( )[ ]txki 222 exp ω−=Ψ
onda che si muove verso x positive ( verso DESTRA )
onda che si muove verso x negative ( verso SINISTRA )
( ) 21, Ψ+Ψ=Ψ tx
questa è la rappresentazione in serie di Fourier
in - 16
Dipartimento di Chimica Prof. Guido Gigli
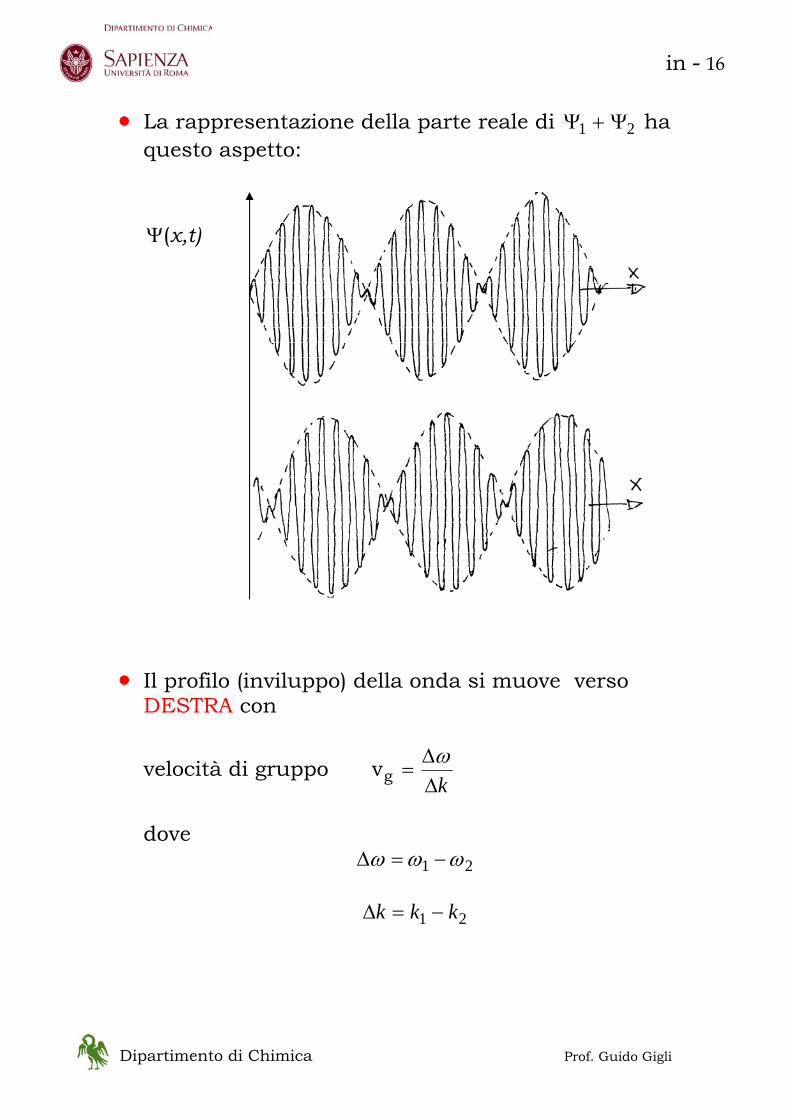
• La rappresentazione della parte reale di 21 Ψ+Ψ ha
questo aspetto:
• Il profilo (inviluppo) della onda si muove verso
DESTRA con
velocità di gruppo kΔ
Δ=
ωgv
dove
21 ωωω −=Δ
21 kkk −=Δ
Ψ(x,t)
in - 17
Dipartimento di Chimica Prof. Guido Gigli
Onda Stazionaria Quando
( ) ( )[ ] ( )
( )
( )tsenitkxekx
eee
eetx
kkk
ti
tiikxikx
tkxitkxi
ωω
ωωω
ω
ω
ωω
−==
+=
+=Ψ
==−==
−
−−
+−−
coscos2cos2
,21
21
f(x) g(t)
quindi indipendentemente da t
▬ ( ) 0cosper 0, ==Ψ kxtx
......,23,2 ππ=kx
▬ i nodi non dipendono dal tempo
in - 18
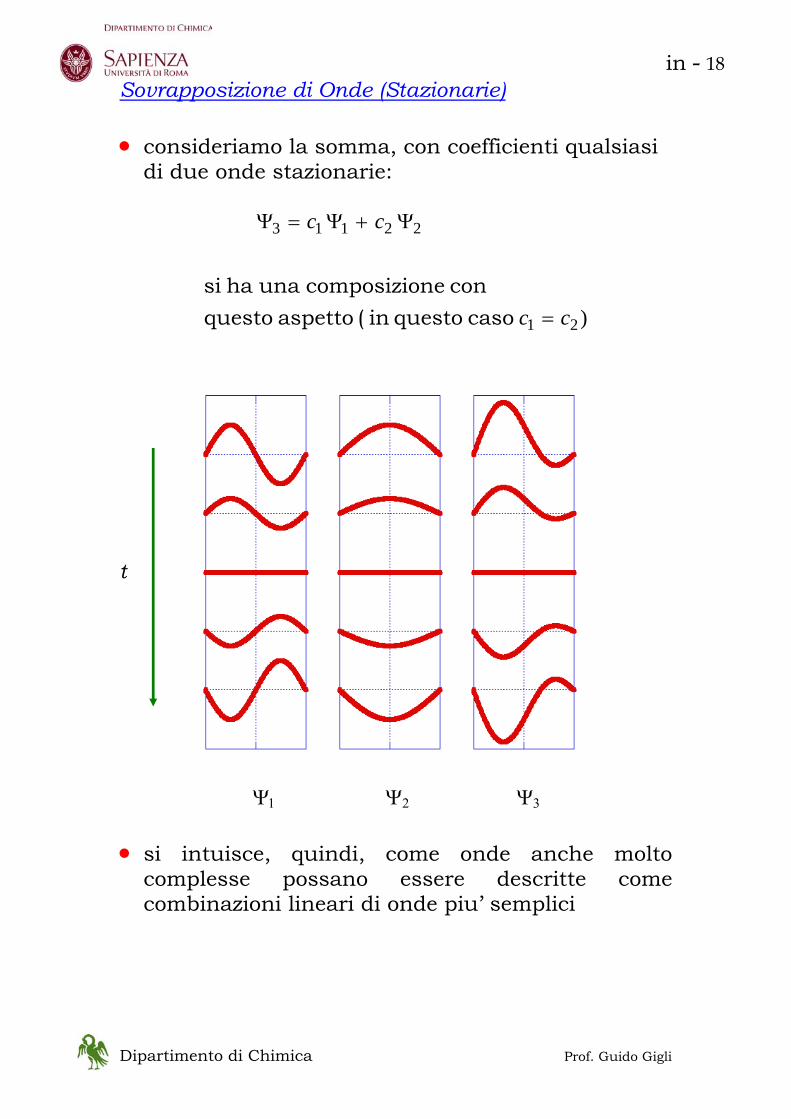
Dipartimento di Chimica Prof. Guido Gigli
Sovrapposizione di Onde (Stazionarie) • consideriamo la somma, con coefficienti qualsiasi
di due onde stazionarie:
)21
22113
cc
cc
=
Ψ+Ψ=Ψ
caso questo in ( aspetto questocon necomposizio una ha si
t Ψ1 Ψ2 Ψ3
• si intuisce, quindi, come onde anche molto
complesse possano essere descritte come combinazioni lineari di onde piu’ semplici
in - 19
Dipartimento di Chimica Prof. Guido Gigli
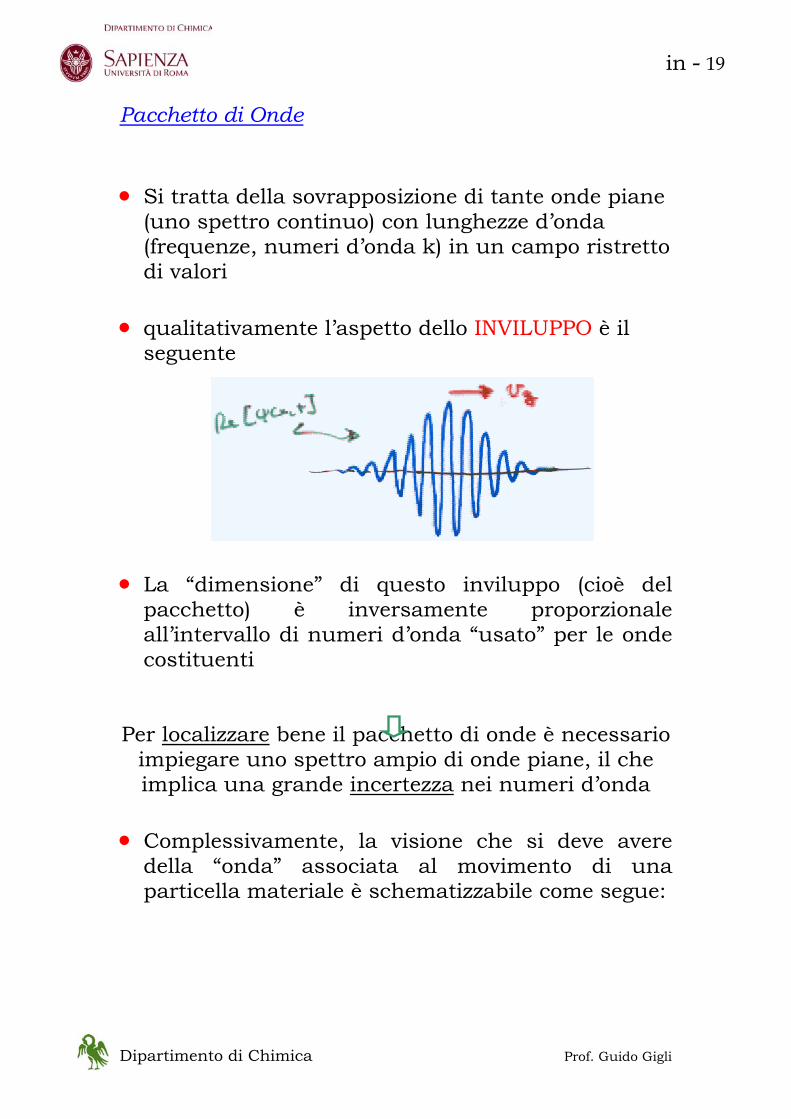
Pacchetto di Onde • Si tratta della sovrapposizione di tante onde piane
(uno spettro continuo) con lunghezze d’onda (frequenze, numeri d’onda k) in un campo ristretto di valori
• qualitativamente l’aspetto dello INVILUPPO è il
seguente • La “dimensione” di questo inviluppo (cioè del
pacchetto) è inversamente proporzionale all’intervallo di numeri d’onda “usato” per le onde costituenti
Per localizzare bene il pacchetto di onde è necessario
impiegare uno spettro ampio di onde piane, il che implica una grande incertezza nei numeri d’onda
• Complessivamente, la visione che si deve avere
della “onda” associata al movimento di una particella materiale è schematizzabile come segue:
in - 20
Dipartimento di Chimica Prof. Guido Gigli
E ben definita ( ν o λ ben definita) E non ben definita ( un intervallo di ν o λ)
una unica onda piana di momento
h=λp un inviluppo di onde piane
non sappiamo dove sia la particella sappiamo abbastanza bene dove sia la particella
• rimane da notare che la velocità di gruppo di
questo pacchetto di onde corrisponde alla velocità classica
Interpretazione fisica dell’onda associata al moto di una particella
Negli esperimenti con doppia fenditura ( alla
YOUNG) le immagini osservate sono dovute all’accumularsi (sia con fotoni che con elettroni) di impatti singoli
La posizione dell’impatto di ogni singola particella non può essere predetta mentre lo è l’effetto cumulativo L’interpretazione degli esperimenti suggerisce di considerare
NON Traiettorie specifiche MA Distribuzioni di probabilità (delle
traiettorie) degli impatti
in - 21
Dipartimento di Chimica Prof. Guido Gigli
• Definiamo la Densità di Probabilità
( )xP ( )dxxP Probabilità che una particella colpisca lo
schermo fra x ed dxx + Rivediamo l’esperimento di Young • Immaginiamo che il moto della particella sia
rappresentato da una funzione d’onda
POSTULIAMO CHE ( ) *2 ψψψ ==xP
• La particella che passa per “a ” è descritta dalla aψ La particella che passa per “b” è descritta dalla bψ
( ) ( ) ( )xxx ba ψψψ += anche questo è postulato perchè la particella non si divide in entità più piccole
caso 1 a Aperta b
Chiusa caso 2 a Chiusa b
Aperta caso 3 a Aperte e b
Chiuse per meta’ tempo
- la funzione Ψ improvvisamente diventa ( “collassa” a ) Ψa
- Pa = ⎪Ψa⎪2
- Ψ collassa a Ψb - Pb = ⎪Ψb⎪2
Pa +Pb = ⎪Ψa⎪2 + ⎪Ψb⎪2
in - 22
Dipartimento di Chimica Prof. Guido Gigli
caso 4 a sempre b Aperte
caso 5 a sempre b
Aperte rivelatore in a
DA NOTARE CHE • L’interpretazione statistica del significato della
funzione d’onda ψ è stata postulata da Born (1926) • Il concetto che la ψ “contiene” tutte le informazioni
sullo stato di moto che rappresenta e “collassa” a stati diversi in una osservazione sperimentale è dovuto ad Heisenberg (1927)
Tutto ciò fa parte della cosiddetta
Interpretazione di Copenhagen
della Meccanica Quantistica
Questa interpretazione è controversa ma, poichè è in accordo con tutte le osservazioni sperimentali di
nostro interesse, è quella che useremo
Pab = = ⎪Ψ⎪2 = ⎪Ψa + Ψb⎪2 = = ⎪Ψa⎪2 + ⎪Ψb⎪2 + Ψa
*Ψb + ΨaΨb*
IA IB IAB termine d’interferenza
La fase di Ψa viene modificata in modo casuale dalla misura in a e, quindi, viene modificato (annullato) il termine di interferenza
in - 23
Dipartimento di Chimica Prof. Guido Gigli
Plausibilità della equazione di Schrödinger • Consideriamo un flusso uniforme di particelle
libere che si muove nel vuoto nella direzione x
questo significa che Il potenziale a cui sono sottoposte è 0V = La funzione d’onda che ne rappresenta una è
DA NOTARE • Non è una “derivazione”. Per esempio dovremmo
ammettere che valga anche quando 0≠V • Qui compare la derivata prima rispetto al tempo
mentre la eq. delle onde classica è
2
2
2
2
v1
tx fase ∂∂
=∂∂ ψψ
• Abbiamo “associato” le relazioni:
mp2
2 2
22
2 xm ∂∂
−h
ti
∂∂
h E
fm - 1
Dipartimento di Chimica Prof. Guido Gigli
FONDAMENTI DELLA MECCANICA QUANTISTICA
Prima delle leggi ( Postulati) vediamo i concetti 1) Variabili dinamiche 2) Funzione d’onda o di stato 3) Operatori
Variabile Dinamica
Si tratta di una qualsiasi variabile che definisce il moto di una particella
posizione, velocità, quantità di moto, energia cinetica, momento angolare, ecc. (il tempo nella formulazione che seguiamo è un
parametro)
Si tratta delle medesime proprieta’ della meccanica classica
Vedremo in seguito che compariranno anche nuove proprieta’ che non hanno corrispondenza in meccanica classica ( SPIN )
La funzione d’onda Nella formulazione che stiamo seguendo (Meccanica
Ondulatoria) è concetto centrale Nell’interpretazione statistica di Born:
( )( ) τdtzyxzyxzyx
dzdydxdzdydxtzyxzyxzyx
nnn
nnnnnn2
222111
1112
222111
,,,,...,,,,,,
...,,,,...,,,,,,
Ψ
Ψ
Dove Ψ = valore assoluto
( ) positivo e reale2/1
←Ψ=ΨΨ∗
fm - 2
Dipartimento di Chimica Prof. Guido Gigli
NoSI
⎢ψ⎪2 dτ è la probabilità che al tempo t si abbia che
particella 1 in x1 ÷ x1+dx1 particella 1 in y1 ÷ y1+dy1 ……………………………………………………… particella n in zn ÷ zn+dzn
• Questo significato fisico della ψ impone che le funzioni
d’onda accettabili siano
“ a buon comportamento “ “ well behaved ”
la probabilità deve essere definita senza ambiguità
1) ad un sol valore sen(x) SI’
dato x f(x) è unico arcsen(x) NO 2) continua con derivata prima continua
lim f(x) = f(a) ( a meno di eccezioni ) x→a
3) finita nel senso di
“ quadrato integrabile “
numero finito l’integrale del quadrato del valore assoluto della funzione deve essere finito
Ndd ∫ =Ψ∫ =ΨΨ ττ 2*
fm - 3
Dipartimento di Chimica Prof. Guido Gigli
NOdxxx
SIdxedxeee xxxx
∞=→
==⋅→
∫
∫∫∞∞−
∞∞−
−∞∞−
−−−
42
21
22
2
2
2
2
2
π
attenzione: il campo di definizione della funzione è
importante
⎩⎨⎧
∞+∞−
=
⎩⎨⎧
=
xxsee
xsee
ix
ix
NO
20
SIπ
θ
• La condizione di “quadrato integrabile” consente la NORMALIZZAZIONE
la particella deve essere da qualche parte
la probabilità su tutto lo spazio deve essere 1
la probabilità “relativa” (integrale finito ) deve poter essere ricondotta alla probabilità assoluta ( 0 ÷ 1 )
la normalizzazione è la divisione per 2/1N
∫ =ΨΨ 12/12/1
*τd
NN
• Si può dimostrare che, una volta normalizzata, la ψ rimane normalizzata col passare del tempo
∫ τΨΨ= dN * non dipende dal tempo
fm - 4
Dipartimento di Chimica Prof. Guido Gigli
• Non tutte le funzioni d’onda possono essere normalizzate→ ⎢ψ⎪2 è la densità di prob. relativa
∫
∫Ψ
Ψ
21
2
21
2
bb
aa
dx
dx
←
per esempio: ( ) ( ) h/Etpxiet,x −=Ψ
Operatori • Si tratta di entità matematiche che trasformano una
funzione f in un’altra funzione g
fg α=
hgf == βαβ ˆˆˆ ( )
ff
f
ff
βα
βα
ααα
ˆˆ
ˆˆ
ˆˆˆ 2
+=
+
=
l’ordine di applicazione degli operatori è importante:
(moltiplicare per) (elevare a) ⋅ f ≠
(elevare a) (moltiplicare per) f per esempio:
αββα
αββα
βα
ˆˆˆˆ
4ˆˆ24ˆˆ
4ˆˆ
≠
⋅=≠⋅=⋅=
⋅==
fffff
Gli operatori α e β non commutano
è la probabilità che sia in a1 ÷ a2 rispetto a quella che sia in b1 ÷b2 b
fm - 5
Dipartimento di Chimica Prof. Guido Gigli
• E’, quindi, importante l’operatore composto
( )
[ ]βα
αββα
ˆ,ˆ
ˆˆˆˆ −
• Se 1ˆˆˆˆ == αββα α e’ il reciproco di β α = β -1
gffg 1ˆˆ −=⇒= αα • Un operatore è LINEARE se
[ ]fkfk
gfgfαα
αααˆˆ
ˆˆˆ=
+=+
• Valgono alcune proprieta
( )( )
[ ] [ ] [ ][ ] [ ] [ ]
( ) ( )γβαγβα
γβαβγαγβα
γαβγβαγβα
γαβαγβα
γβγαγβα
ˆˆˆˆˆˆ
ˆ,ˆˆˆˆ,ˆˆ,ˆˆ
ˆ,ˆˆˆˆ,ˆˆˆ,ˆ
ˆˆˆˆˆˆˆ
ˆˆˆˆˆˆˆ
=
+=
+=
+=+
+=+
COMMUTATORE
e’ una entità centrale a tutto lo sviluppo della meccanica quantistica
d/dx e’ lineare exp non sono lineari √
fm - 6
Dipartimento di Chimica Prof. Guido Gigli
Autofunzioni ed Autovalori • In generale
fg α= dove g ed f sono
Ψ=Φ α linearmente indipendenti
( )dipendenti elinearment0...2211 ⇒=+++ nn fcfcfc
quando per una particolare funzione f1
111ˆ faf =α autofunzione di α
autovalore corrispondente (numero complesso)
N.B.: in questo caso g = a1f1 ed f1 sono lin. dipendenti c1f1 + c2f2 =0 -1⋅g + a1f1 = -a1f1 + a1f1 = 0 • Di norma esistono molte autofunzioni di un operatore
iii aψψα =ˆ
Il sistema descritto da una di queste ψi è in un AUTOSTATO per l’operatore α
• Vi possono essere più autofunzioni con il medesimo
autovalore
si ha DEGENERAZIONE per esempio: più autofunzioni con la medesima energia
totale ↔ più stati di moto con la stessa energia
ogni autofunzione di α è unica nel senso di essere linearmente indipendente dalle altre
fm - 7
Dipartimento di Chimica Prof. Guido Gigli
• Le autofunzioni possono essere infinite e costituire un insieme continuo oppure discreto
Prodotto Scalare ed Ortogonalità
• La definizione di prodotto scalare è:
( ) ( ) ( ) ( )
( ) ( )∫ ∫ ∫
∫∫∫∫
∞
∞+
∞−
ΨΦ
ΨΦΨΦ
π πτ
ϕθθϕθϕθ20 0 0
2*
**
sin,,,,
,,,,
dddrrrr
dxdydzzyxzyxdxxx
in forma più compatta ∫ τΨΦ d*
oppure usando la NOTAZIONE di DIRAC
ψΦ
bra(c)ket ∫ ΨΦ τd*
si possono immaginare queste corrispondenze ψΦ ( a sinistra della barretta: complesso coniugato)
*ΦΨ=ΨΦ
quindi e’ anche REALE
kxkx keedxd
= nxnnxdxd sinsin 2
2
2−=
ΨΦ=ΨΦ *kk
ΨΦ=ΨΦ kk
*ΨΨ=ΨΨ
fm - 8
Dipartimento di Chimica Prof. Guido Gigli
• Due funzioni sono ortogonali se
0=ΨΦ oppure se, per un insieme (“set”) di autofunzioni di un operatore α jiglituttiperji ≠⇒=ΨΨ 0
Se poi sono anche normalizzate sono ortonormali quando
⎪⎩
⎪⎨⎧
≠→=
=→=⇒=ΨΨ
jiper
jiper
ij
ijijji 0
1
δ
δδ
Aggiunto di un operatore
• L’aggiunto di un operatore lineare α è quell’operatore
α + per il quale (per ogni Ψ e Φ)
ΨΨ=ΨΨ
ΨΦ=ΨΦ
ΨΦ=ΨΦ
+
+
+
αα
αα
αα
ˆˆ
ˆˆ
ˆˆ
Operatori Hermitiani • Un operatore α è Hermitiano se α + = α (cioè è
autoaggiunto)
ΨΨ=ΨΨΨΦ=ΨΦ αααα ˆˆˆˆ quindi
all’interno di un integrale un operatore hermitiano opera indifferentemente a destra od a sinistra
δij delta di Kronecker
fm - 9
Dipartimento di Chimica Prof. Guido Gigli
• Alternativamente si puo’ definire un operatore α come
Hermitiano se per un insieme di funzioni Ψi:
( ) τατα
αα
ααα
dd jiij
jiij
ijjiij
∫∫ ΨΨ=ΨΨ
⎪⎩
⎪⎨
⎧
ΨΨ=ΨΨ
ΨΨ=ΨΨ=ΨΨ
**
*
*
ˆˆ
ˆˆ
ˆˆˆ
• L’Hermitianità di un operatore è una proprietà dell’operatore stesso, delle funzioni e del campo di integrazione
• Vediamo qualche esempio
per α = d/dx dovrebbe valere che
ΨΦ=ΨΦ αα ˆˆ cioe’
ΨΦ=ΨΦ dxd
dxd
dxdx dxd
dxd ΨΦ=ΨΦ ∫∫ **
Invece integrando per parti il primo membro, si ha:
[ ] ΨΦ−=ΨΦ−ΨΦ=ΨΦ=ΨΦ ∫∫∞+∞− dx
ddxd
dxd
dxd dxdx ***
è la cosiddetta regola del “turn over”
0
fm - 10
Dipartimento di Chimica Prof. Guido Gigli
• ancora per esempio
( ) [ ] ( )
ΨΦ−=ΨΦ
=ΨΦ−−ΨΦ−=Ψ−Φ=Ψ−Φ
∫
∫∫∞+∞−
dxd
dxd
dxd
dxd
dxd
idxi
dxiidxii
hh
hhhh
*
***
• anticipiamo ora che tutti gli operatori utili della
meccanica quantistica posseggono entrambe queste caratteristiche:
sono Lineari ed Hermitiani
• se per α e β lineari ed hermitiani vale che [α , β ]=0
(cioè commutano )
allora anche il loro prodotto α β
è lineare ed hermitiano
dxd
idxdi h
h =−=αper
0
fm - 11
Dipartimento di Chimica Prof. Guido Gigli
POSTULATI
I Postulato Lo stato di un sistema fisico è definito da una funzione d’ onda ψ delle coordinate e del tempo. Tutte le informazioni sullo stato del sistema sono contenute in questa funzione
• Esiste una ψ (qr ,t ) che deve essere : - ad un sol valore - continua - quadrato
integrabile
• Di norma assumeremo che la ψ sia
normalizzata
1=ΨΨ • La denominazione “ vettore di stato” ci
introduce allo Spazio di Hilbert e notazione di Dirac
• Si tratta di uno schema concettuale alternativo per le funzioni d’onda quantomeccaniche
Funzione di Stato Funzione d’onda Vettore di Stato
fm - 12
Dipartimento di Chimica Prof. Guido Gigli
• In analogia allo spazio cartesiano definito da 3 vettori ortogonali
un insieme di funzioni ortogonali (ortonormali) possono essere considerate come i vettori di base di uno spazio vettoriale (di Hilbert)
nella notazione di Dirac i vettori base dello spazio di
Hilbert sono chiamati “Kets”
ioppurekets iΨ⇒ alcune caratteristiche:
- ii ck Ψ=Ψ e’ un nuovo ket nella stessa direzione ma di grandezza diversa
- quando un operatore agisce su di un ket il
risultato e’ un altro ket iii Ψ=Ψ=Φ αα ˆˆ
• Poichè le ψi sono funzioni complesse lo spazio dei Ket è
complesso e si introducono i complessi coniugati (trasposti) "Bra”
ioppurebra iΨ⇒
N.B. ++
+
Ψ=ΨΨ=Ψ
ΨΨΨ
iiii
iii
e
infatti
ket del aggiuntol' è
fm - 13
Dipartimento di Chimica Prof. Guido Gigli
• Rivediamo il prodotto scalare
ψψ Prodotto interno
è un numero reale positivo in analogia al prodotto di un numero complesso per il suo complesso coniugato
• Il prodotto scalare di un Bra <Ψj⏐ e di un Ket ⎟α ψi>
viene scritto ijij αα ˆˆ oppure ΨΨ
ijij Aij oppure oppure αα ˆˆ ΨΨ
II Postulato
Ad ogni osservabile fisica è associato un operatore lineare ed Hermitiano
• In generale ad ogni funzione classica della posizione e
del momento
( )prA rr,=
corrisponde un operatore lineare Hermitiano
( )∇− hr irA ,
• Siamo usando la cosiddetta
“ rappresentazione delle posizioni”
fm - 14
Dipartimento di Chimica Prof. Guido Gigli
xip
xx
x ∂∂
−⇒
⋅⇒
h
• Esiste anche la
“ rappresentazione dei momenti”
⋅⇒∂∂
+⇒
xx
x
ppp
ix h
Vediamo il dettaglio delle regole
• Alle variabili dinamiche classiche vengono associati
degli operatori mediante
regole postulate
Variabile
Operatore
x ⋅x
y ⋅y
z ⋅z px
xi
xi ∂∂
−=∂∂
hh
py y
iyi ∂
∂−=
∂∂
hh
pz z
izi ∂
∂−=
∂∂
hh
• Questi operatori vengono usati nelle espressioni
classiche
tutti Hermitiani
fm - 15
Dipartimento di Chimica Prof. Guido Gigli
( )( )
2
222
22
xxi
xip
xxxx
x∂∂
−=⎟⎠⎞
⎜⎝⎛
∂∂
−⋅⎟⎠⎞
⎜⎝⎛
∂∂
−⇒
⋅=⋅⋅⇒
hhh
Energia Cinetica
( ) ( )
Δ−
=∇−
=⎟⎟⎠
⎞⎜⎜⎝
⎛
∂∂
+∂∂
+∂∂−
=
++=++==
m2m2zyxm2T
pppm21vvvm
21TE
22
2
2
2
2
2
2
22
2z
2y
2x
2z
2y
2xcin
hhhˆ
“ Nabla quadro”
Energia Potenziale
( )( )
⎪⎩
⎪⎨⎧
=
=→
⎩⎨⎧
→=
221cost
fisico problema dal dipende
,,,,,
kxV
VV
zyxVtzyxV
VEpot
VV =)
Energia totale
TVTVEEHE
TVEEE
cinpottotale
cinpottotale
ˆˆˆˆˆˆˆ +=+=+==
+=+=
↑ è lineare ed Hermitiano Momento Angolare
momento angolare classico prLM rrrr∧==
Rotatore Rigido
Oscillatore Armonico
fm - 16
Dipartimento di Chimica Prof. Guido Gigli
zyx pppzyxkji
pr
rrr
rr ∧
( ) ( ) ( )xyzxyz ypxpkxpzpjzpypiL −+−+−=
rrrr
xL yL zL
⎥⎦
⎤⎢⎣
⎡⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−∂∂
+⎟⎠⎞
⎜⎝⎛
∂∂
−∂∂
+⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−∂∂
−=x
yy
xz
xx
zy
zz
yiL hˆ
xL yL zL
III Postulato In un sistema gli unici valori che una variabile dinamica può assumere sono gli autovalori dell’ operatore corrispondente
iai
aa
i
iii
iii
=
Ψ=Ψ
Ψ=Ψ
α
αα
ˆ
ˆˆ
cinetica energia della valorii sono T dove T T iiii Ψ=Ψ
• In altri termini - Se il sistema è in un autostato dell’operatore ogni singola misura della variabile dinamica corrispondente ci fornirà come risultato esattamente uno degli autovalori
α operatore associato alla variabile dinamica, per esempio:
fm - 17
Dipartimento di Chimica Prof. Guido Gigli
- Se il sistema non è in un autostato, ma è descritto da una generica ψ, ogni volta che si fa una misura esso “finisce” in uno degli autostati
per citare Dirac
“Una misura fa sempre saltare il sistema in un autostato della variabile dinamica che si misura”
IV Postulato
Il valore medio di una serie di misure della variabile α su di un insieme di sistemi ciascuno dei quali è esattamente nel medesimo stato ψ è fornito dalla relazione
τα
ααα
da ∫ ΨΨ=
ΨΨ=ΨΨ=∗ ˆ
ˆˆ
N.B. altre denominazioni: valore atteso
attendibile di aspettazione
per esempio:
Ψ⎟⎟⎠
⎞⎜⎜⎝
⎛+∇−Ψ=Ψ+∇−Ψ=
ΨΨ=ΨΨ=
ΨΨ=ΨΨ=
Vm
Vm
E
ppp
xxx
xxx
22
22
22
ˆˆ
ˆˆ
hh
• Da notare che ψ può non essere autofunzione di α
fm - 18
Dipartimento di Chimica Prof. Guido Gigli
• Da notare che ogni singola misura fornirà come
risultato uno degli autovalori ai dell’operatore α per esempio:
10243 4321 aaaa +++
=α
naturalmente perchè il valore di <α> sia “attendibile” il numero di misure deve essere grande
V Postulato la dipendenza temporale della funzione di stato ψ è fornita dall’equazione differenziale
tiH
∂Ψ∂
=Ψ hˆ
Equazione di Schroedinger dipendente dal tempo
• Per una particella
( )t
itzyxVm ∂
Ψ∂=Ψ⎥
⎦
⎤⎢⎣
⎡+∇− h
h ,,,2
22
Altri Postulati
vedremo che saranno necessari ulteriori postulati per trattare lo
SPIN
fm - 19
Dipartimento di Chimica Prof. Guido Gigli
• Per il momento vediamo una serie di conseguenze ( dimostrabili) di questi postulati che chiameremo corollari
corollario 1 Le quantità misurabili sono numeri reali corollario 2a Le autofunzioni ψm e ψn dell’operatore α con
autovalori am ed an ( cioè con autovalori diversi) sono ortogonali
2b Due autofunzioni degeneri di un operatore α (cioè con il medesimo autovalore) possono essere combinate linearmente per realizzare funzioni ortogonali ed ancora autofunzioni dell’operatore (questa proprietà discende dalla linearità degli operatori di interesse in meccanica quantistica)
corollario 3 Se l’energia potenziale di un sistema non dipende esplicitamente dal tempo la densità di probabilità non dipende dal tempo e l’energia totale è ricavabile dall’equazione
( ) ( )nEnH 33ˆ Ψ=Ψ
Equazione di Schroedinger indipendente dal tempo
• Di quest’ultimo vediamone il perchè per una particella
fm - 20
Dipartimento di Chimica Prof. Guido Gigli
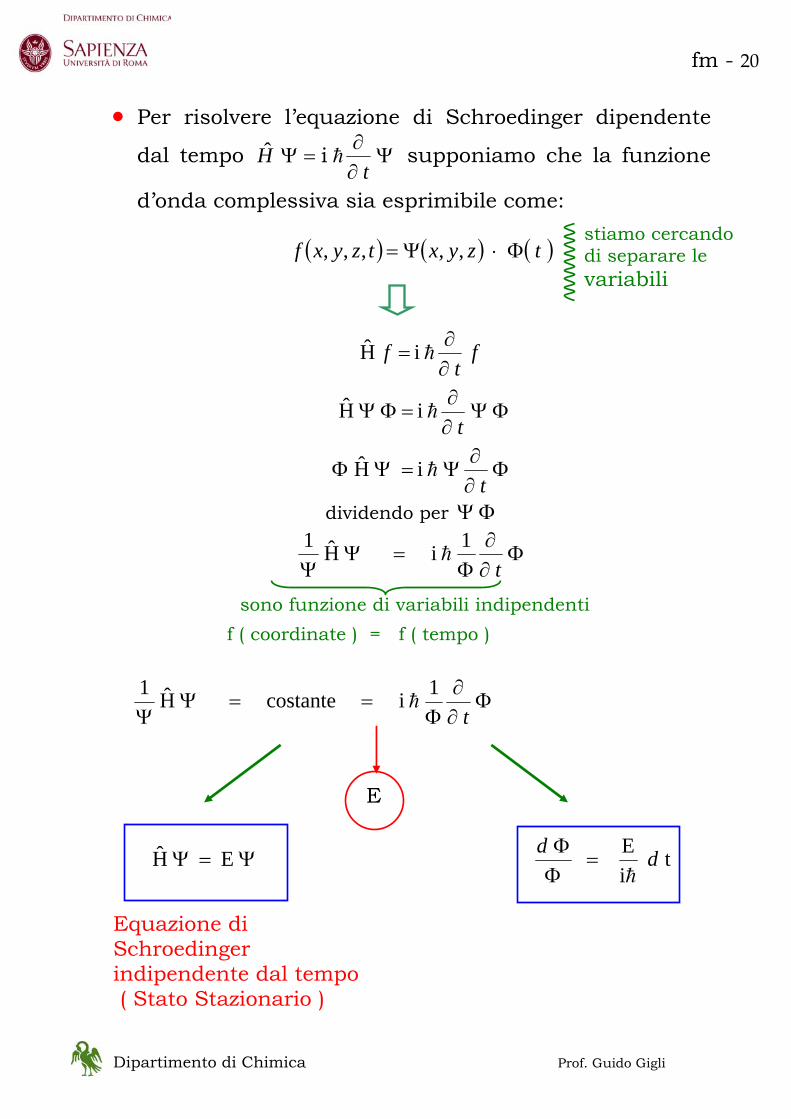
• Per risolvere l’equazione di Schroedinger dipendente
dal tempo Ψ∂∂
=Ψt
H h i ˆ supponiamo che la funzione
d’onda complessiva sia esprimibile come:
( ) ( ) ( )tzyxtzyxf Φ⋅Ψ= ,,,,,
ft
f∂∂
= h i H
ΦΨ∂∂
=ΦΨt
h i H
Φ∂∂
Ψ=ΨΦt
h i H
dividendo per ΦΨ
Φ∂∂
Φ=Ψ
Ψ t1 i H1
h
sono funzione di variabili indipendenti f ( coordinate ) = f ( tempo )
Φ∂∂
Φ==Ψ
Ψ t1 i costante H1
h
E
Ψ=Ψ E H t iE ddh
=ΦΦ
Equazione di Schroedinger indipendente dal tempo ( Stato Stazionario )
stiamo cercando di separare le variabili
fm - 21
Dipartimento di Chimica Prof. Guido Gigli
• Vediamo la dipendenza temporale
( )tiEt
iE
AeAet
dtiEd
hh
h
−==Φ
=ΦΦ
• Complessivamente
( ) ( ) ( )tiE
Aezyxtzyxf h−
Ψ=ΦΨ= ,,,,
⎜f⎟2=ff*= ψψ* non dipende dal tempo • Più in generale
( ) ( )∑−
Ψ=
Ψ=Ψ
i
tiiE
ii
iii
ezyxctzyxf
EH
h,,,,,
ˆ
tutte le soluzioni sono esprimibili come somma di stati stazionari
• Il tutto può essere così rappresentato
- tenendo conto che
h
hhh
/ E con ..ecc. :tra oscilla
i=
−=−
ν111
sincos
i,,,-i,-
tEitEe iitiiE
- la funzione d’onda oscilla da ampiezze positive ad ampiezze negative ecc. con frequenza proporzionale all’Energia
A=1 per avere ƒ normalizzata A inglobato nella costante di normalizzazione di Ψ(x,y,z)
ci costanti arbitrarie complesse
Re Ψ
Imm Ψ
fm - 22
Dipartimento di Chimica Prof. Guido Gigli
• se immaginiamo di osservare la sola parte reale, nel caso di uno stato stazionario, la “onda” oscilla nel tempo senza spostarsi nello spazio
………………………….. Corollario 4 E’ possibile conoscere
contemporaneamente e con precisione i valori di due variabili dinamiche soltanto se gli operatori corrispondenti commutano
INFATTI
Se entrambe le variabili devono essere note con precisione deve esistere una funzione di stato ψ per la quale
Ψ=Ψ=Ψ=Ψ⇒Ψ=Ψ abaaa ββαβα ˆˆˆˆ)
Ψ=Ψ=Ψ=Ψ⇒Ψ=Ψ babbb ααβαβ ˆˆˆˆˆ
QUINDI
[ ]βααββααββα ˆ,ˆ0ˆˆˆˆˆˆˆˆ ==−⇒Ψ=Ψ
VICEVERSA abbiamo già visto che non tutti gli operatori della quantomeccanica commutano
Ψ∂∂
=Ψx
xpx x i- hˆˆ
Ψ∂∂
Ψ=Ψ∂∂
=Ψx
xiixx
ixpx - - - hhh )(ˆˆ
fm - 23
Dipartimento di Chimica Prof. Guido Gigli
Ψ=Ψ− hixppx xx )ˆˆˆˆ( [ ] hipx x =ˆ,ˆ
• tutto ciò differisce dalla meccanica classica • si parla di
Grandezze CONIUGATE (variabili) NON CONIUGATE
A tutto ciò è legato il
Principio di indeterminazione di Heisenberg
Per un sistema in uno stato arbitrario ψ le generiche osservabili fisiche α e β non possono essere determinate simultaneamente con precisione se α e β non commutano
• Una misura della imprecisione di α e β è la varianza dei risultati a e b
• Si può dimostrare che
[ ][ ]βασσ
βασσ
αββασσ
ˆ,ˆ21
ˆ,ˆ21
ˆˆˆˆ21
≥
ΨΨ≥
Ψ−Ψ≥
ba
ba
ba
• Se α e β commutano si possono conoscere con precisione entrambe le variabili
• Se α e β non commutano non si può scendere al di sotto del limite precedente
il commutatore di x e xp vale hi
fm - 24
Dipartimento di Chimica Prof. Guido Gigli
• per α ≡ x e β ≡ xp)
( )
2
221
21 2/1
h
hhhh
≥
=⋅−=≥
xpx
xpx iii
σσ
σσ
• Poichè è sempre possibile conoscere con precisione una delle variabili, l’altra è determinata nei limiti indicati
• Vediamo le proprietà di commutazione e,quindi,le
eventuali limitazioni nella misura dei momenti angolari
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−∂∂
−=y
zz
yiLx hˆ
⎟⎠⎞
⎜⎝⎛
∂∂
−∂∂
−=z
xx
ziLy hˆ
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−∂∂
−=x
yy
xiLz hˆ
• quanto vale il commutatore di xL ed yL ?
⎟⎟⎠
⎞⎜⎜⎝
⎛
∂∂∂
+∂∂
∂−
∂∂
−∂∂
∂+
∂∂
−=
=⎟⎠⎞
⎜⎝⎛
∂∂
−∂∂
⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−∂∂
−=
zyxz
xyz
zxy
xzyz
xy
zx
xz
yz
zyLL yx
222
2
222
2ˆˆ
h
h
⎟⎟⎠
⎞⎜⎜⎝
⎛
∂∂∂
+∂∂
+∂∂
−∂∂
∂−
∂∂∂
−=zy
xzy
xz
xyxy
zxz
yzLL xy
2
2
222
22ˆˆ h
zxyyx Liy
xx
yLLLL ˆˆˆˆˆ 2 hh =⎟⎟⎠
⎞⎜⎜⎝
⎛∂∂
−∂∂
−=−
fm - 25
Dipartimento di Chimica Prof. Guido Gigli
• generalizzando
xyzzy LiLLLL ˆˆˆˆˆ h=−
yzxxz LiLLLL ˆˆˆˆˆ h=−
• Invece per il Momento angolare totale
22222 ˆˆˆˆˆ LLLLL zyx =++=
0ˆˆˆˆ 22 ≡− xx LLLL in conclusione
• è possibile conoscere il momento angolare totale ed una delle componenti
• non è possibile conoscere il momento angolare totale e due componenti
• Esiste anche una limitazione relativa alla energia ed al
tempo che,peraltro,è di natura diversa. Per vederla sarebbe necessario affrontare il problema di
come varia nel tempo il valore di aspettazione
e la conseguente Indeterminazione Energia-Tempo • Il tempo non è una variabile simile a
posizione,momento,energia: è un Parametro
- nella nostra rappresentazione (quella di Schroedinger) non esiste un “operatore” tempo
- la incertezza in tempo non è esprimibile come un intervallo dei valori di aspettazione
fm - 26
Dipartimento di Chimica Prof. Guido Gigli
- per una generica variabile cui sia associato l’operatore
β si puo’ dimostrare che
2h
≥ΔtEσ
è l’intervallo di tempo nel quale il valore di aspettazione di β
cambia di una quantità pari alla sua deviazione standard
l’incertezza sull’energia dipende dalla natura della variabile associata a β . Se cambia poco nel tempo l’incertezza su E sarà piccola e viceversa
• il tutto puo’, piu’ semplicemente, essere espresso
dicendo che se tΔ è il tempo di vita di un determinato stato di un sistema allora la indeterminazione della energia di questo stato è tale che
2h
≥ΔtEσ
• in linea di principio, per uno stato stazionario ∞=Δt e la energia è nota con esattezza
• Il caso di maggior interesse è quello della Spettroscopia nella quale si misura una differenza di energia corrispondente ad una frequenza
νhE =Δ 2/h≅Δ=Δ thtE νσσ
πσν 4/1≅Δt
• In pratica tΔ dipende dal tempo di permanenza nello stato eccitato ( 10-8 ÷ 10-10 s )
113~197 1031031010 −−−− ⋅÷⋅≅÷≅ cms νν σσ
fm - 27
Dipartimento di Chimica Prof. Guido Gigli
Corollario 5 Le autofunzioni di ogni operatore quantomeccanico corrispondente ad una variabile osservabile costituiscono un insieme completo e ortonormale.
Cosa è un insieme di funzioni “completo”? Per quanto ci serve è sufficiente la definizione: Una serie di funzioni ,..., 21 ΦΦ con determinati requisiti è completa se una generica funzione f con gli stessi requisiti è esprimibile come
∑ Φ=i
iicf
CONSEGUENZE: • ogni combinazione lineare di un insieme di
autofunzioni degeneri è ancora una autofunzione con il medesimo autovalore
• Il corollario ci consente di dedurre l’effetto di un
operatore su di una funzione che non è una delle sue autofunzioni
Indaghiamo meglio sulla natura del valore di aspettazione di un operatore
lo stato del sistema è descritto da una Ψ
α ha un insieme completo e ortogonale di autofunzioni iΦ
La funzione f può essere “espansa” in termini delle funzioni iΦ
sistema fisico
normalizzata non autofunzione di un operatore α
fm - 28
Dipartimento di Chimica Prof. Guido Gigli
Dal corollario si ha che ∑=Ψi
iic φ
ed il valore di aspettazione della variabile associata ad α
è
∑ ∑∑ ∑
∑ ∑∑ ∑
∑∑
∗∗
∗∗
=ΦΦ=
=ΦΦ=ΦΦ=
ΦΦ=ΨΨ=
i jijjji
i jjijji
i jjjiji
i jjiji
jjj
iii
accacc
acccc
cca
δ
α
αα
ˆ
ˆˆ
∑∑ == ∗
iii
iiii acacca 2
- Ogni singola misura della variabile
corrispondente allo operatore α fornirà uno degli autovalori ia
- Il valore medio delle misure è una media pesata sui ii cc * degli autovalori ai
- Ogni 2ic è la “probabilità” che venga misurato il
corrispondente autovalore ia
- 2ic è la probabilità che il sistema venga trovato
in uno stato descritto dalla funzione iΦ Peraltro
jiji
ii
iijj
ii
ccc
c
=ΦΦ=ΦΦ=ΨΦ
ΨΦ=
∑∑
: infatti
• Tutto ciò può essere considerato come: - L’espansione della ψ nelle autofunzioni iΦ
- Lo stato Ψ come SOVRAPPOSIZIONE degli AUTOSTATI iΦ (uno stato è scomposto nei vettori componenti che sono stati “puri”)
fm - 29
Dipartimento di Chimica Prof. Guido Gigli
Corollario 6 • Se due operatori α e β hanno un insieme completo di
autofunzioni simultanee allora i due operatori commutano
• Se due operatori α e β commutano possiamo trovare un insieme completo di funzioni che sono autofunzioni simultaneamente di entrambi gli operatori
Principio di corrispondenza di Bohr • una delle forme in cui puo’ essere espresso è:
“ Per h che tende a zero, cioè per dimensioni macroscopiche del sistema in esame, i risultati della trattazione quantomeccanica tendono a divenire uguali a quelli della analisi classica”
(naturalmente rimane vero che poichè h non è, in realta’, una variabile i risultati classici e quantistici sono diversi)
• alternativamente ( per esempio): “ Un pacchetto d’onde ben definito si muove in accordo alle leggi di meccanica classica della particella che rappresenta”
INFATTI secondo il teorema di Ehrenfest
dtxd
mp = FdxVd
dtpd
=−=
si tratta della generalizzazione quantomeccanica delle espressioni classiche del moto; cioè, i valori di
aspettazione di F ed dtVd ,p seguono le leggi classiche:
Newton di e amFdt
xdmp ⋅==
fm - 30
Dipartimento di Chimica Prof. Guido Gigli
Un breve approfondimento sui “bra” e “ket” • ΨΨ ce rappresentano il medesimo stato fisico
(per postulato) – in termini di vettori questo significa che ne conta soltanto l’orientazione e non la lunghezza
• iΨ è un membro dello spazio “duale” dei “bra” –
“corrisponde” al ket iΨ
• i*c Ψ è il bra duale di ic Ψ
• gli operatori agiscono sui bra da destra αΨ ˆ e si genera un altro bra
• Ψα in generale non corrisponde a αΨ ˆ SE
Ψα corrisponde a +αΨ ˆ
α ed +α sono l’uno l’aggiunto dell’altro SE
α = +α α è hermitiano ed autoaggiunto
• αΨ ˆ ed Ψα non hanno senso (non sono leciti)
• ( ) ( )Ψα•Ψ ˆ è il prodotto scalare interno ΨαΨ ˆ • esiste anche il Prodotto esterno ΓΦ che è un operatore ΓΦ che quando opera sulla Ψ la trasforma in una funzione (un ket)
proporzionale alla Φ infatti
ΓΦ Ψ = ΨΓΦ = tetancosΦ
fm - 31
Dipartimento di Chimica Prof. Guido Gigli
• un particolare prodotto esterno è l’operatore di
proiezione iii P=ΦΦ
• quando è applicato ad una Ψ : ΨΦΦ ii
il risultato è un ket proporzionale a iΦ cioè
l’operatore iP ha proiettato Ψ in iΦ
• nel caso dell’espansione di una Ψ in un set di funzioni ortonormali iΦ sappiamo che:
Ψ = ∑ Φi
iic
e che ΨΦ= iic
quindi Ψ = ∑ ΦΨΦ
iii = ∑ ΨΦΦ
iii
poichè poi la Ψ è arbitraria ne consegue che 1=ΦΦ∑
iii
relazione di completezza
perché è legata al criterio di completezza • la relazione precedente puo’ essere inserita in una
equazione in qualsiasi punto della medesima: per esempio
1
1
22 ==ΨΦ=ΨΦΦΨ
=Ψ⎟⎠
⎞⎜⎝
⎛ΦΦΨ==ΨΨ
∑∑∑
∑
ii
iii
ii
iii
c
Università di Roma “ La Sapienza” ps - 1
Dipartimento di Chimica Prof. Guido Gigli
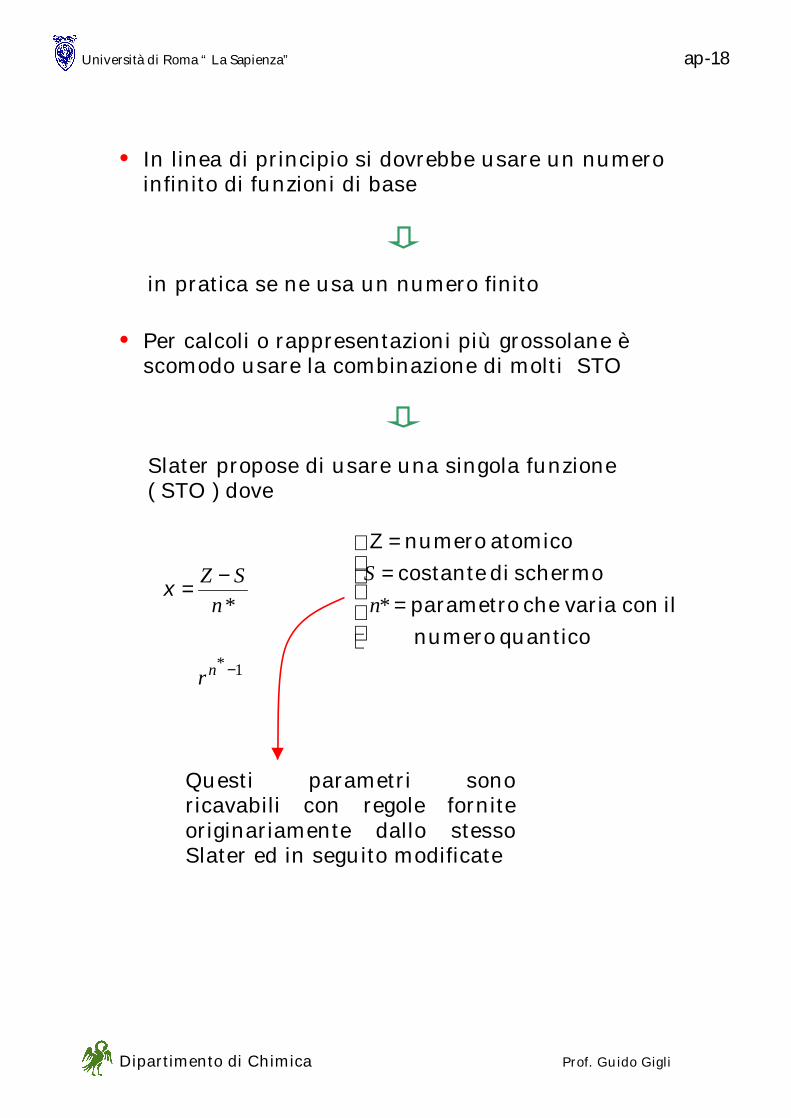
PARTICELLA NELLA SCATOLA
• Iniziamo ad affrontare i sistemi “modello” che sono utili in Chimica (e per i quali si riesce a risolvere la equazione di Schroedinger) con un modello adatto ai GAS IDEALI
- particelle puntiformi - nessuna interazione fra le particelle
0=
TdV
dU
- soltanto energia cinetica
in una dimensione
esplicitiamo le condizioni di un sistema fisico nel quale una particella di massa m è confinata in una buca di potenziale di profondità infinita
V(x) ha questo aspetto
( ) axxV ≤≤= 00
( )ax
xxV
><
∞=0
• ( ) ( )tfxV ≠ usiamo la eq. di Schroedinger non
dipendente dal tempo
Ψ=Ψ EH
( ) Ψ=Ψ
+− ExV
dx
d
m 2
22
2
h
0 a x
V=∞∞∞∞ V=∞∞∞∞
V(x)
Università di Roma “ La Sapienza” ps - 2
Dipartimento di Chimica Prof. Guido Gigli
per ( ) ∞=xV
( )
01
2
2
2
2
2
2
22
=Ψ⇒Ψ
∞=Ψ⇒
⇒Ψ∞=Ψ⇒Ψ∞=Ψ∞−=Ψ−
dx
d
dx
dE
dx
d
m
h
La probabilità ( )2ψ di trovare la particella fuori
dalla scatola è nulla per ( ) 0=xV
Ψ=Ψ−⇒Ψ=Ψ−22
2
2
22 2
2 h
h Em
dx
dE
dx
d
m
kpem
pE h==
2
2
Ψ=Ψ− 22
2
kdx
d
Le soluzioni di questa eq. differenziale devono essere funzioni proporzionali alla loro derivata seconda come
f f ’ f”
kxsin kxk cos kxk sin2−
kxcos kxk sin− kxk cos2−
ikxe ikxeik ikxeki 22
ikxe− ikxeik −− ikxeki −22
• Usiamo la soluzione generale
( ) kxBkxAx cossen +=Ψ
A e B costanti arbitrarie da determinare con le CONDIZIONI al CONTORNO
in questo caso sono meno semplici da
usare
Università di Roma “ La Sapienza” ps - 3
Dipartimento di Chimica Prof. Guido Gigli
( ) ( ) ( )( ) ( )
( ) ( )
) sen)sen(- soluzioni nuove fornisce non 0 (n
) ovunque 0 (x) rebbe significhe 0 n (
n1,2,...,n con
) ovunque 0 (x) ( eaccettabil non
ax
0x
:ementeconseguent
a xe per
scatola dalla fuori
θθ
π
−=<=Ψ=
==⇒==Ψ⇒=
=
==Ψ⇒==+==Ψ⇒=
===Ψ⇔=Ψ
nkaka
AkaA
kaAa
BBA
x
0sin
00sin
sin0
0cos0sin00
00
0
le condizioni al contorno
hanno introdotto la QUANTIZZAZIONE con n
Dal punto di vista fisico soltanto per alcuni valori dell’onda associata (alla De Broglie) la funzione d’onda rappresenta correttamente il moto
πnka =
a
hnhp
n
a
na
nn
n
2
2
2
==
=
=
λ
λ
πλπ
m
p
ma
hnE
namE
n 28
2
2
2
22
21
2
==
=
πh
Università di Roma “ La Sapienza” ps - 4
Dipartimento di Chimica Prof. Guido Gigli
Energia
• L’energia è quantizzata
• Il più basso possibile livello è 18 2
2
1 =⇒= nma
hE
c’è una Energia di Punto zero
In accordo con il principio di indeterminazione di
Heisenberg Infatti
ax =∆ la particella e’ da qualche parte nella scatola
xx pp 2=∆ il suo momento varia nel
campo [ ]xxx ppp +−⊆ ,
hna
nhapapx xx =
⋅=⋅⋅=∆∆2
22
• L’energia e le differenze di energia 2
11
ae
m∝
per m d’ordine di grandezza macroscopico ed a si ha un CONTINUO
limite classico Principio di CORRISPONDENZA Funzione d’onda ( ) kxAxn sin=Ψ
xa
nA
πsin=
A viene determinato con la condizione di
Normalizzazione
Università di Roma “ La Sapienza” ps - 5
Dipartimento di Chimica Prof. Guido Gigli
1sin022 =
∫a dxx
a
nA
π
usando
θθ
θθθ
θθ
2cos1sin2
2cossincos
1sincos
2
22
22
−=
=−
=+
=→==→=
⇒=⇒=πθ
θθ
ππθ
nax
xd
n
adxx
a
n 00
( ) [ ] [ ]
( ){ }
aA
aA
nn
aA
n
aAd
n
aA nnn
2
12
002
2sin2
1
22cos1
222
00
2
0
2
=
==−−=
−=−∫
ππ
θθπ
θπ
θ πππ
( ) xa
n
axn
πsin
2 2
1
=Ψ
• Possiamo controllare che queste autofunzioni siano ortogonali
mnnm ≠⇒=ΨΨ 0
come esempio prendiamo in considerazione i primi due livelli 21 == nn
Università di Roma “ La Sapienza” ps - 6
Dipartimento di Chimica Prof. Guido Gigli
∫ =⋅a
dxxa
xa
A0
2 2sinsin
ππ ααα cos22 sensen =
∫
∫
=
⋅=
=⋅⋅
a
a
xa
dxa
aA
dxxa
xa
xa
A
0
22
0
2
sinsin2
cossinsin2
πππ
πππ
0sin0
3 =
∝a
xa
π
• vediamo l’aspetto di queste autofunzioni
• La funzione d’onda completa per gli stati stazionari
( ) ( )t
ma
hniiEt
exa
n
aextx hh
28
22
sin2
,−−
⋅
⋅=⋅Ψ=Ψ π
Università di Roma “ La Sapienza” ps - 7
Dipartimento di Chimica Prof. Guido Gigli
Osservazioni
• All’aumentare di n la probabilità di trovare la particella tende sempre di più ad essere uniforme in tutta la scatola
• Le Ψ con n pari ed n dispari hanno simmetria diversa
n pari antisimmetriche n dispari simmetriche
si tratta di una conseguenza generale della simmetria del Potenziale
PARITA’ delle autofunzioni di un sistema con
Potenziale SIMMETRICO
• La energia cresce al crescere del numero di NODI delle autofunzioni
Una semplice interpretazione fisica è che
tanto più la Ψ “curva” tanto più aumenta la Energia Cinetica
PERCHE’
l’operatore T implica l’uso di una derivata seconda
Ψ2
anche qui è all’opera il principio di
corrispondenza
n
grande
Università di Roma “ La Sapienza” ps - 8
Dipartimento di Chimica Prof. Guido Gigli
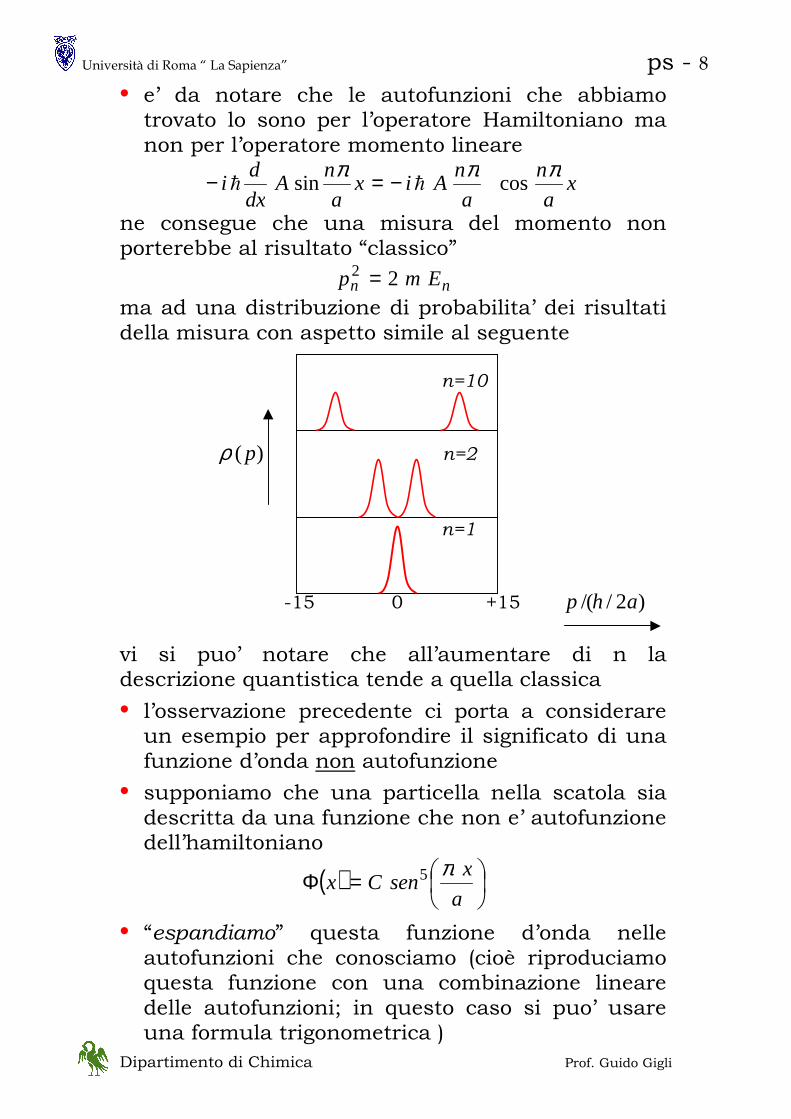
• e’ da notare che le autofunzioni che abbiamo trovato lo sono per l’operatore Hamiltoniano ma non per l’operatore momento lineare
xa
n
a
nAix
a
nA
dx
di
πππcossin hh −=−
ne consegue che una misura del momento non porterebbe al risultato “classico”
nn Emp 22 =
ma ad una distribuzione di probabilita’ dei risultati della misura con aspetto simile al seguente n=10 )( pρ n=2
n=1 -15 0 +15 )2/(/ ahp
vi si puo’ notare che all’aumentare di n la descrizione quantistica tende a quella classica
• l’osservazione precedente ci porta a considerare un esempio per approfondire il significato di una funzione d’onda non autofunzione
• supponiamo che una particella nella scatola sia descritta da una funzione che non e’ autofunzione dell’hamiltoniano
( )
=Φa
xsenCx
π5
• “espandiamo” questa funzione d’onda nelle autofunzioni che conosciamo (cioè riproduciamo questa funzione con una combinazione lineare delle autofunzioni; in questo caso si puo’ usare una formula trigonometrica )
Università di Roma “ La Sapienza” ps - 9
Dipartimento di Chimica Prof. Guido Gigli
la misura della energia di questa particella darebbe come risultato uno (ed uno soltanto) dei tre valori E1, E3, E5 (il sistema collassa in uno degli autostati) con queste probabilita’
794.01510
10
8 222
2
12
2
1 =++
=⇐= Pam
hE
198.01510
5
8
9222
2
12
2
3 =++
=⇐= Pam
hE
008.01510
1
8
25222
2
12
2
5 =++
=⇐= Pam
hE
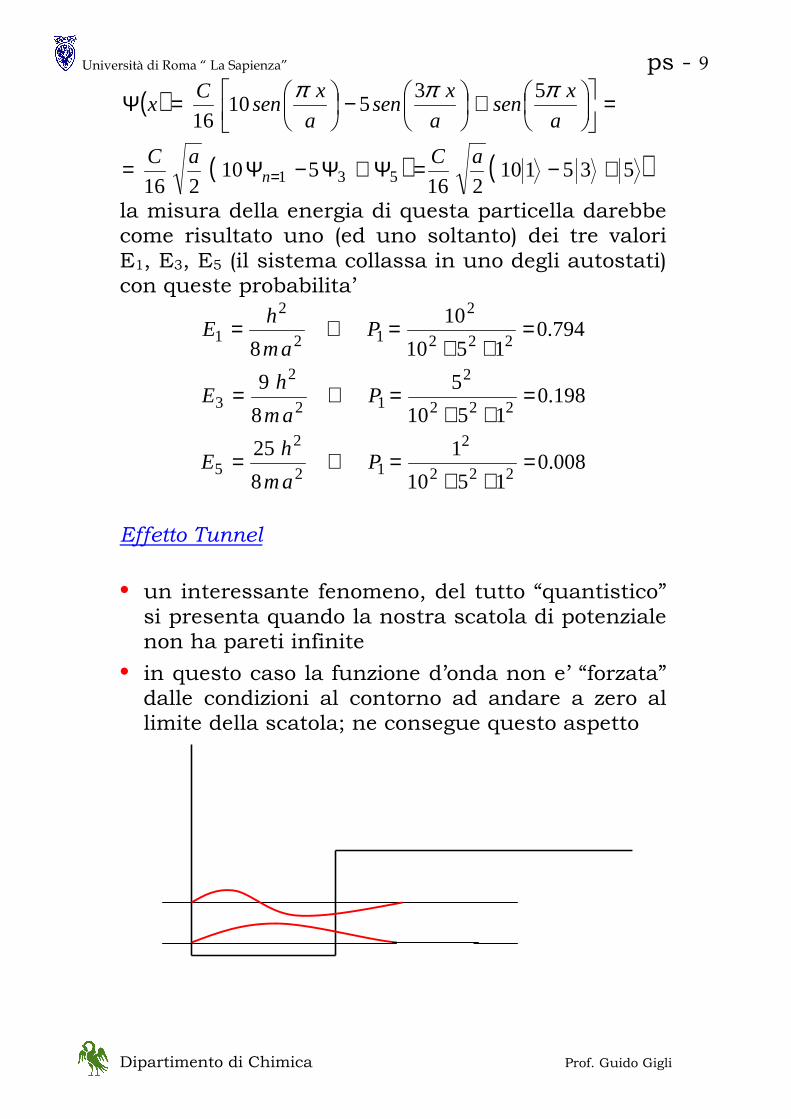
Effetto Tunnel
• un interessante fenomeno, del tutto “quantistico” si presenta quando la nostra scatola di potenziale non ha pareti infinite
• in questo caso la funzione d’onda non e’ “forzata” dalle condizioni al contorno ad andare a zero al limite della scatola; ne consegue questo aspetto
( )
( ) ( )535110216
510216
53510
16
531 +−=Ψ+Ψ−Ψ=
=
+
−
=Ψ
=aCaC
a
xsen
a
xsen
a
xsen
Cx
n
πππ
Università di Roma “ La Sapienza” ps - 10
Dipartimento di Chimica Prof. Guido Gigli
• la particella puo’ trovarsi al di fuori della scatola laddove il potenziale e’ maggiore della sua energia totale
T = E – V < 0 il che è, classicamente, un assurdo
• se due scatole di questo tipo sono affiancate
• la particella puo’ essere “trovata” sia nella scatola di destra che in quella di sinistra
• in termini classici la particella passa da sinistra a destra senza superare la barriera di energia potenziale ( la “collina” ); è come se ci fosse un “tunnel” fra le due scatole
• questo effetto dipende da : altezza della barriera (diminuisce con) larghezza delle scatole (diminuisce con) massa della particella (diminuisce con)
• in sintesi, al limite classico (grande barriera, ampie scatole, grandi masse) l’effetto sparisce
• alcune reazioni che coinvolgono atomi leggeri come l’idrogeno risentono fortemente di questo effetto tunnel
Università di Roma “ La Sapienza” ps - 11
Dipartimento di Chimica Prof. Guido Gigli
Particella in una scatola tridimensionale
• Il risultato ottenuto può essere facilmente esteso al caso a 3 dimensioni
( )( ) zyxdivalorealtroqualsiasiperzyxV
czbyaxzyxV
,,,,
0000,,
⇒∞=≤≤≤≤≤≤⇒=
( ) ( )zyxEzyxzyxm
,,,,2 2
2
2
2
2
22
Ψ=Ψ
∂∂+
∂∂+
∂∂− h
• Usiamo il metodo della separazione delle variabili
( ) ( ) ( ) ( )
Ez
Z
Zmy
Y
Ymx
X
Xm
zZyYxXzyx
=∂∂−
∂∂−
∂∂−
⋅⋅=Ψ
2
22
2
22
2
22 1
2
1
2
1
2
,,
hhh
x, y e z sono indipendenti e quindi
zyx Edz
Zd
ZmE
dy
Yd
YmE
dx
Xd
Xm=−=−=−
2
22
2
22
2
22 1
2
1
2
1
2
hhh
( )
( )
( )2
22
2
22
2
22
8sin
2
8sin
2
8sin
2
mc
hnEz
c
n
czZ
mb
hnEy
b
n
byY
ma
hnEx
a
n
axX
EEEE
zz
z
yy
y
xx
x
zyx
=⇒
⋅=
=⇒
⋅=
=⇒
⋅=
++=
π
π
π
• In questo caso compare la DEGENERAZIONE ( che qui e’ accidentale )
SE cba ==
Università di Roma “ La Sapienza” ps - 12
Dipartimento di Chimica Prof. Guido Gigli
( )2222
2
,,8
zyxznynxn nnnma
hE ++=
nx ny nz 22 ma8hE Degenerazione
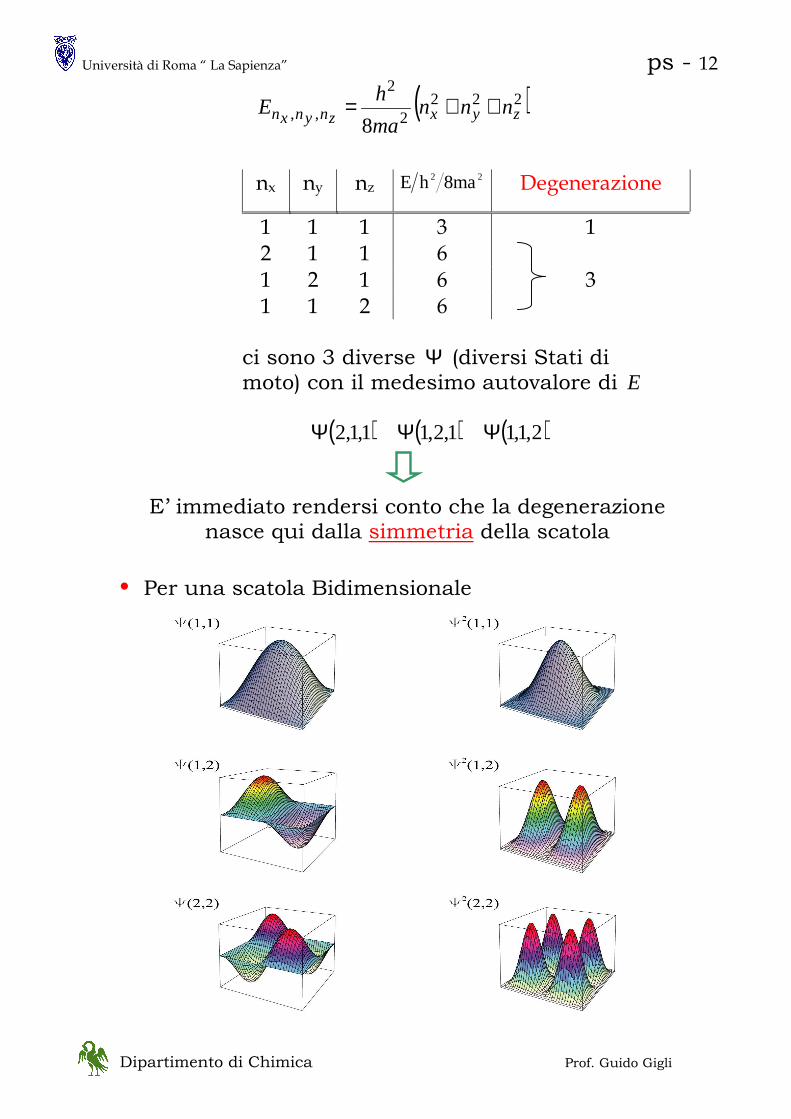
1 1 1 3 1 2 1 1 6 1 2 1 6 3 1 1 2 6
ci sono 3 diverse Ψ (diversi Stati di moto) con il medesimo autovalore di E
( ) ( ) ( )2,1,11,2,11,1,2 ΨΨΨ
E’ immediato rendersi conto che la degenerazione nasce qui dalla simmetria della scatola
• Per una scatola Bidimensionale
Università di Roma “ La Sapienza” oa - 1
Dipartimento di Chimica Prof. Guido Gigli
OSCILLATORE ARMONICO
• Sistema modello delle vibrazioni delle molecole (approssimazione della realtà)
• Vediamolo da un punto di vista Classico e Quantistico
Secondo la Meccanica Classica
xkfxfrrrr
−=⇒∝
2
2
2 cost2
1
dt
xdmkxxmf
kxdxfVdx
dVf
=−⇒=
+=−=⇒−= ∫
&&
( )2
1
2
1
massima eelongazion
2cos
=
=
⇒−=
m
k
a
tax
πν
φνπ
( ) mkemk 221 ωω ==
( ) ( ) ( )φνπφνπνπ −+−=
=+=+=
tkatam
kxxmVTEtot
2cos2
12sin2
2
12
1
2
1
222222
22&
( ) 2222
21
...cos...sin21
akakEtot =+=
• da notare 1. E è continua 2. ν è legata alle proprietà del sistema m e k
Università di Roma “ La Sapienza” oa - 2
Dipartimento di Chimica Prof. Guido Gigli
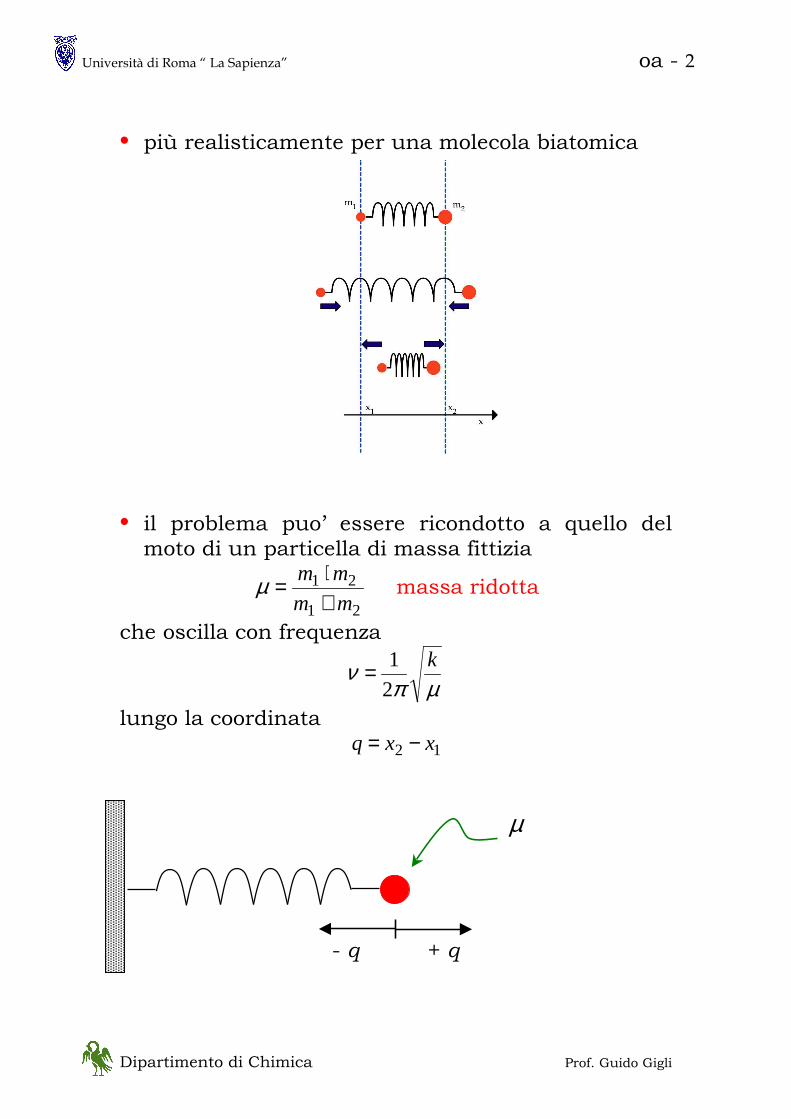
• più realisticamente per una molecola biatomica
• il problema puo’ essere ricondotto a quello del moto di un particella di massa fittizia
21
21
mm
mm
+⋅=µ massa ridotta
che oscilla con frequenza
µπν k
2
1=
lungo la coordinata
12 xxq −=
µ
- q + q
Università di Roma “ La Sapienza” oa - 3
Dipartimento di Chimica Prof. Guido Gigli
Secondo la Meccanica Quantistica
• Possiamo usare la eq. di Schroedinger NON dipendente dal tempo ( )[ ]tfV ≠
( ) ( )22
222
2
1
22
1
2
1
33ˆ
xmm
pkxmvVT
nEnH
xx ω+=+=+
Ψ=Ψ
222
22
2
1
2ˆ
ˆˆ
xmdx
d
mH
xxedx
dipx
ω+−=
⋅=−=
h
h
• Introduciamo una nuova variabile adimensionale
222
1
2
1
xm
ydxm
dyxm
yhhh
ωωω =⇒
=⇒
=
22
222
2
22
222
1
2ˆ y
dy
dy
mm
dy
dm
mH
ωωω
ωω hhh
h
h +−=+−=
( ) ( ) ( )yEyyydy
d Ψ=Ψ+Ψ− 22
2
22
ωω hh
( ) ( )yEydy
dy Ψ=Ψ
−
2
22
2
ωh
• si tratta, anche qui come nel caso della particella nella scatola, di una equazione differenziale di II ordine che va risolta
la soluzione è abbastanza complicata
• per il momento vediamone i soli risultati
Università di Roma “ La Sapienza” oa - 4
Dipartimento di Chimica Prof. Guido Gigli
• la funzione d’onda è
( ) ( ) ( ) 2
2
2
2 y
n
y
n eyHceyucy−−
⋅=⋅=Ψ
( )yHn⋅ è un polinomio detto di Hermite il quale
dipende dal numero n che è un intero positivo
• in effetti, anche qui come nel caso della particella nella scatola, sono le condizioni imposte alla funzione d’onda ( in questo caso di andare a zero all’infinito per consentire di essere quadrato integrabile ) che rendono il numero n quantizzato
• i polinomi sono esprimibili in forma compatta con una unica formula: ( formula di RODRIGUEZ )
( ) ( )nyn
ny
n edy
decyH 1-ccon =
⋅= − 22
• vediamo, per esempio, i primi polinomi ( )
( ) ( )
( ) ( )( )( ) 124816
128
242442
22
1
244
33
222222222
222211
0
+−=
−=
−=−=
+−=
=
−−=
⋅=
=
−−−
−−
yyyH
yyyH
yyeeeyeeyH
yyeeedy
deyH
yH
yyyyy
yyyy1-
Università di Roma “ La Sapienza” oa - 5
Dipartimento di Chimica Prof. Guido Gigli
• esistono anche delle relazioni che consentono di ricavare automaticamente un polinomio dagli adiacenti
( ) nnn HHnHy +−= −− 21 122
• anche gli autovalori della energia dipendono dal numero quantico n e quindi per ogni n
dove
( )
classica frequenza dove ⇐
==
+=
+=+=
21
2
12
2
1
2
112
2
1
m
ke
hnnnEn
πνπνω
νωω hh
• Torniamo alla soluzione generale del problema dell’oscillatore armonico
( ) ( ) ( ) 2
2
2
2 y
n
y
n eyHceyucy−−
⋅=⋅=Ψ
Normalizzando la ψ si ha
( ) ( ) ( )
( )x
kx
eyHn
yy
nn
n
21
4121
2
2
4121!2
1
hh
mmydove =
=
=Ψ−
ω
π
( ) ( )yEyH nnn Ψ=Ψˆ
Università di Roma “ La Sapienza” oa - 6
Dipartimento di Chimica Prof. Guido Gigli
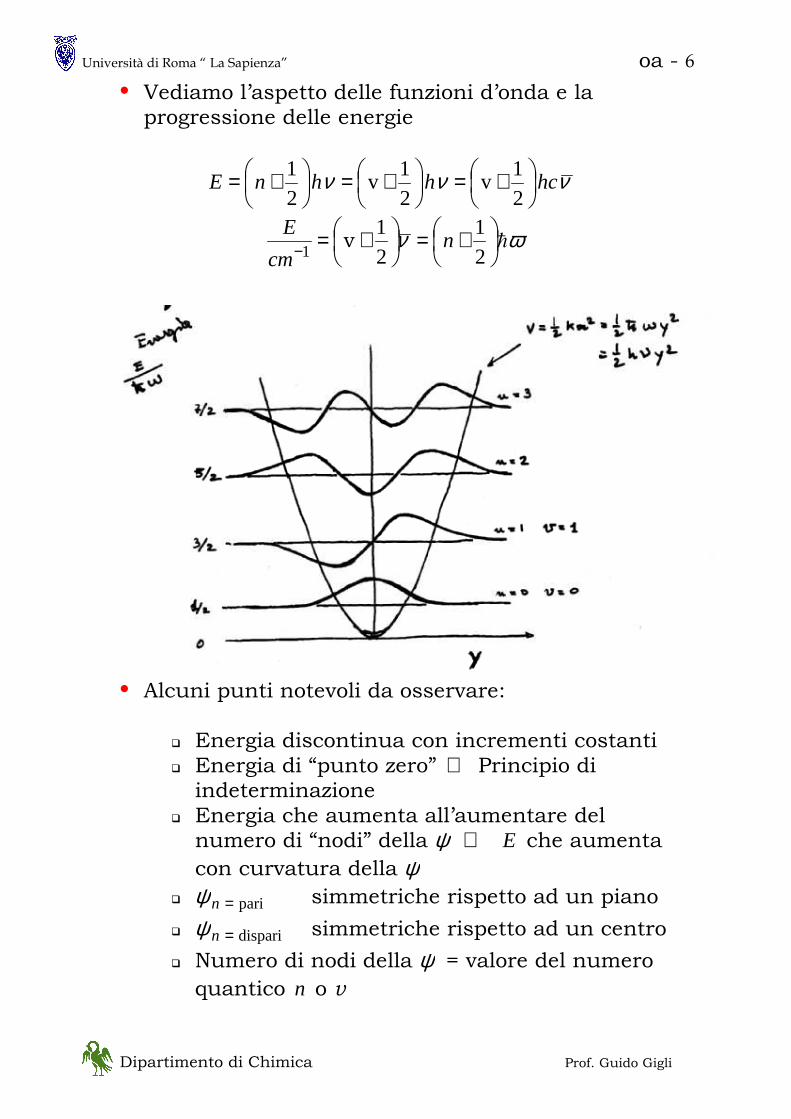
• Vediamo l’aspetto delle funzioni d’onda e la progressione delle energie
ννν hchhnE
+=
+=
+=21
v21
v21
ων h
+=
+=− 21
21
1n
cm
Ev
• Alcuni punti notevoli da osservare:
� Energia discontinua con incrementi costanti � Energia di “punto zero” ⇔ Principio di indeterminazione
� Energia che aumenta all’aumentare del numero di “nodi” della E⇔ψ che aumenta
con curvatura della ψ � pari=nψ simmetriche rispetto ad un piano
� dispari=nψ simmetriche rispetto ad un centro
� Numero di nodi della ψ = valore del numero quantico n o v
Università di Roma “ La Sapienza” oa - 7
Dipartimento di Chimica Prof. Guido Gigli
• Per quanto riguarda la densità di probabilità
ψψ *
� La particella si può trovare al di fuori dei limiti della elongazione massima classica; anche qui abbiamo lo
EFFETTO TUNNEL ( )21−∝ mν
la penetrazione nella barriera cresce al diminuire della massa perché, a parita’ di potenziale, una massa piccola significa una frequenza grande ed una energia di punto zero elevata � La densità di probabilità tende a quella classica per ∞→n
Università di Roma “ La Sapienza” oa - 8
Dipartimento di Chimica Prof. Guido Gigli
Metodo degli Operatori per la soluzione della equazione di Schroedinger
• è istruttivo vedere questo metodo, essenzialmente algebrico, che sfrutta le proprieta’ di commutazione degli operatori e non richiede di affrontare complicate elaborazioni matematiche
Autovalori
• Abbiamo visto che la eq. di Schroedinger, con una nuova variabile adimensionale è diventata
( ) ( )yEyH Ψ=Ψˆ
con l’operatore
−=
2
22
2ˆ
dy
dyH
ωh
• oservando l’hamiltoniano si puo’ immaginare che per analogia con
( ) ( ) ( )bababa −+=− 22
si possa cercare un modo per fattorizzare l’operatore e, quindi, ridurre la equazione differenziale dal II al I ordine
• in effetti questo è possibile con i nuovi operatori
Hermitiani non
−
=
−=
+
=
+=
+
ωωα
ωωα
m
pix
m
dy
dy
m
pix
m
dy
dy
ˆ
22
1ˆ
ˆ
22
1ˆ
2
1
2
1
h
h
ricaviamo il commutatore di questi operatori:
+≠ αα ˆˆ
Università di Roma “ La Sapienza” oa - 9
Dipartimento di Chimica Prof. Guido Gigli
−−=
+−=
−++−=
−⋅
+=
+
+
12
1ˆˆ
121
121
21
ˆˆ
2
22
2
22
2
22
dy
dy
dy
dy
dy
d
dy
dy
dy
dyy
dy
dy
dy
dy
αα
αα
[ ] [ ] 1ˆ,ˆ1ˆ,ˆ −== ++ αααα
ωωαα hh
+=
+= +21ˆ
21
ˆˆˆ NH
αα ˆˆˆ +≡N Operatore Numero
• La relazione precedente ci dice che: NedH)) differiscono - del fattore ωh
-della costante1/2 QUINDI
NedH)) -commutano
-hanno le medesime autofunzioni
iiiiii
iii
ENH
N
Ψ=Ψ
+=Ψ
+=Ψ
Ψ=Ψ
ωνω
ν
hh2
1
2
1ˆˆ
ˆ
ων h
+=2
1iiE
infine, poiché si dimostra che gli autovalori
dell’operatore N sono i soli numeri interi positivi (incluso lo zero):
ni =ν νω hnnE
+=
+=2
1
2
1h
Università di Roma “ La Sapienza” oa - 10
Dipartimento di Chimica Prof. Guido Gigli
• Con i commutatori
[ ] [ ] ++ +=−= αααα ˆˆ,ˆˆˆ,ˆ NeN
si puo’ vedere che gli strani operatori +αα ˆˆ ed che
abbiamo introdotto fanno scendere e salire nella scala dei valori permessi di energia
α) è un operatore che fa “scendere” lungo la scala dei valori permessi di energia
[ ] iiii NNN Ψ−=Ψ−Ψ=Ψ αααα ˆˆˆˆˆˆ,ˆ
iiiiii NN Ψ−Ψ=Ψ−Ψ=Ψ αναααα ˆˆˆˆˆˆˆ
( ) iiiN Ψ−=Ψ ανα ˆ1ˆˆ
Cosa significa questa relazione?
• iψα è una autofunzione di N con autovalore ( )1i −ν
• α agendo sull’autostato iψ (con autovalore iν )
dell’operatore N lo modifica in un altro autostato con autovalore più basso 1i −ν
• L’energia dello stato in accordo con
( ) ων h21+= iiE
viene diminuita di ωh
QUINDI
α viene chiamato OPERATORE di DISCESA STEP DOWN (DISTRUZIONE) (ANNICHILAZIONE)
Università di Roma “ La Sapienza” oa - 11
Dipartimento di Chimica Prof. Guido Gigli
ANALOGAMENTE
( ) iiiN ψανψα ++ += ˆ1ˆˆ
+α viene chiamato OPERATORE di SALITA
STEP UP (CREAZIONE)
Autofunzioni
• Schematicamente
+
≡Ψ
≡Ψ++ 1n
1-nautovalori con N di niautofunzio sono
n
n ˆˆˆ
ˆˆ
n
n
αα
αα
QUINDI
nα è una funzione Φ che soddisfa la equazione ( )Φ−=Φ 1ˆ nN
CIOE’
Φ è la ( ) 111 1ˆ −−− −=Ψ⇒ nnn nN ψψ
• In generale 1cosˆ −= ntnα
Dalle condizioni di normalizzazione
1ˆ −= nnnα
11ˆ ++=+ nnnα
In altri termini:
Con gli operatori α ed +α possiamo ottenere qualsiasi funzione d’onda dalla successiva o dalla
precedente
Università di Roma “ La Sapienza” oa - 12
Dipartimento di Chimica Prof. Guido Gigli
Ci rimane da determinare la forma analitica delle
autofunzioni
• Vediamo la prima della serie
0
02
10
2
1
00ˆˆ
2410
200
0
0
0
==Ψ
=Ψ⇒Ψ−=Ψ
≠=Ψ
+
=≡Ψ
−2y
-
2y-
e :ottiene si ndonormalizza
e cost
:ha si poichè e
π
αα
ydy
d
dy
dy
• Ora possiamo ottenere le altre autofunzioni “SCALANDO” la serie con l’operatore di salita
( )yHydoveyy
yy
dy
dy
124
1
24
1
24
1
0
24
1
2222
1
2
2
2
1
2
10ˆ110
==
=
+Ψ=
=
−==+
−−
−
−+
2y-
2y-
2y-
2y-
ee
e
e
ππ
π
πα
Università di Roma “ La Sapienza” oa - 13
Dipartimento di Chimica Prof. Guido Gigli
=
−=
−==+ −+ 22y-eydy
dy
dy
dy 41212
2
11
2
11ˆ211 πα
=
+−
=−−
−
−
22y-22y-
22y-
ee
e
2
222
22
41214121
4121
21
yy
yy
ππ
π
( ) 224141212412121 122222
2y-
22y-22y- eee −=
−
= −−−− yy πππ
( ) 224121 1222
2y-e−= −− yπ
( )yH2
IN CONCLUSIONE (ritornando alla variabile x)
xm
y21
=h
ω
( ) ( ) ( ) 2241
21!2 yn
nn eyH
mnx −−
=Ψhπω
Regole di Selezione
• In spettroscopia le “Regole di Selezione”, cioè le regole che ci dicono quali sono le transizioni permesse, dipendono, sostanzialmente, da:
Integrale del momento di transizione
mnmn d φµφτφµφ∫ =*
In sostanza sviluppando il momento di Dipolo Elettrico compaiono degli integrali del tipo
mxn ˆ
Università di Roma “ La Sapienza” oa - 14
Dipartimento di Chimica Prof. Guido Gigli
• Vediamo il valore di questi Elementi di Matrice
• Usiamo la definizione degli operatori α e +α per ricavare x
( )
−
=
+
=⇐
=++
+
ωωα
ωωα
ωαα
m
pix
m
m
pix
m
xm
x
x
ˆˆ
2ˆ
ˆˆ
2ˆ
ˆ22
ˆˆ21
21
21
h
h
h
( )ααω
ˆˆ2
ˆ21
+
= +
mx
h
• peraltro gli operatori di salita e discesa fanno “salire e scendere” lungo la scala delle
autofunzioni n
1ˆ1ˆ ++=−= + nnnn 1ne n αα
• Abbiamo nxnmxn ˆ'ˆ ≡
1,'
1,'
11'1ˆ'
1'1
1'01'ˆ'
++
−
+=++=
−=⇒=−≠⇒=
=−=
nn
nn
nnnnnn
nn
nnnnnnnn
δα
δα
QUINDI
[ ]
[ ]1,'1,'
21
21
12
ˆ'ˆ'2
ˆ'
−+
+
++
=
+
=
nnnn nnm
nnnnm
nxn
δδω
ααω
h
h
Università di Roma “ La Sapienza” oa - 15
Dipartimento di Chimica Prof. Guido Gigli
( )
=−
+
=+
nm
nxn
nm
nxn
21
21
2ˆ1
12
ˆ1
ω
ω
h
h
−≠+≠
=1'
1'0ˆ'
nn
nnpernxn
E’ come dire che abbiamo:
La regola delle possibili transizioni
REGOLA di SELEZIONE
1±=∆n
L’energia corrispondente è
νω hE ==∆ h la transizione avviene
alla frequenza ν dell’oscillatore classico
m
K
π2
1
Indeterminazione
( ) ( )222 xxxxx −≡−=σ
( ) ( ) 222xxxxpx pppp −≡−=σ
• Per valutare queste quantità ci servono i valori di aspettazione di x e xp per un oscillatore armonico
nel generico autostato n
Università di Roma “ La Sapienza” oa - 16
Dipartimento di Chimica Prof. Guido Gigli
• per x 0ˆ =nxn
• per xp
( ) =−
= + nm
innpn ααωˆˆ
2'ˆ'
21h
[ ]=−
= + nnnnm
i ααωˆ'ˆ'
2
21h
( )[ ]1,'21
1,'21
21
12 −+ −+
= nnnn nnm
i δδωh
0ˆ =npn
QUINDI
( ) 22222 0 xpxx pxx ==−= σσ
SI RICAVA CHE
( )21ˆ2 += nm
nxnωh
( )21ˆ 2 += nmnpn x ωh
IN CONCLUSIONE
( ) ( ) ( )
( ) h
hh
21
2121 21212121
+=
=++
=
n
nmnmxpx
ωω
σσ
per n = 0
2
h=xpx σσ
Esattamente il limite minimo del principio di
Heisenberg
Università di Roma “ La Sapienza” oa - 17
Dipartimento di Chimica Prof. Guido Gigli
Valori medi di Energia cinetica e potenziale
• Facendo uso dei risultati di 2x e 2
xp possiamo
ricavare i valori medi della Energia cinetica, T, e della Energia potenziale V
+=
+===
+=
+===
2
1
2
1
2
1
222
1
21
21
21
21
21
22
22
22
nnm
mx
knxknV
nnmm
pm
nm
pnT
ωω
ω
ωω
hh
hh
L’energia si ripartisce per metà in
Energia Cinetica e per metà in
Energia Potenziale
VTE +=
In accordo con il Teorema del VIRIALE secondo il quale
- per un sistema sottoposto ad un potenziale
nrV ∝
VnT2
1=
Università di Roma “ La Sapienza” oa - 18
Dipartimento di Chimica Prof. Guido Gigli
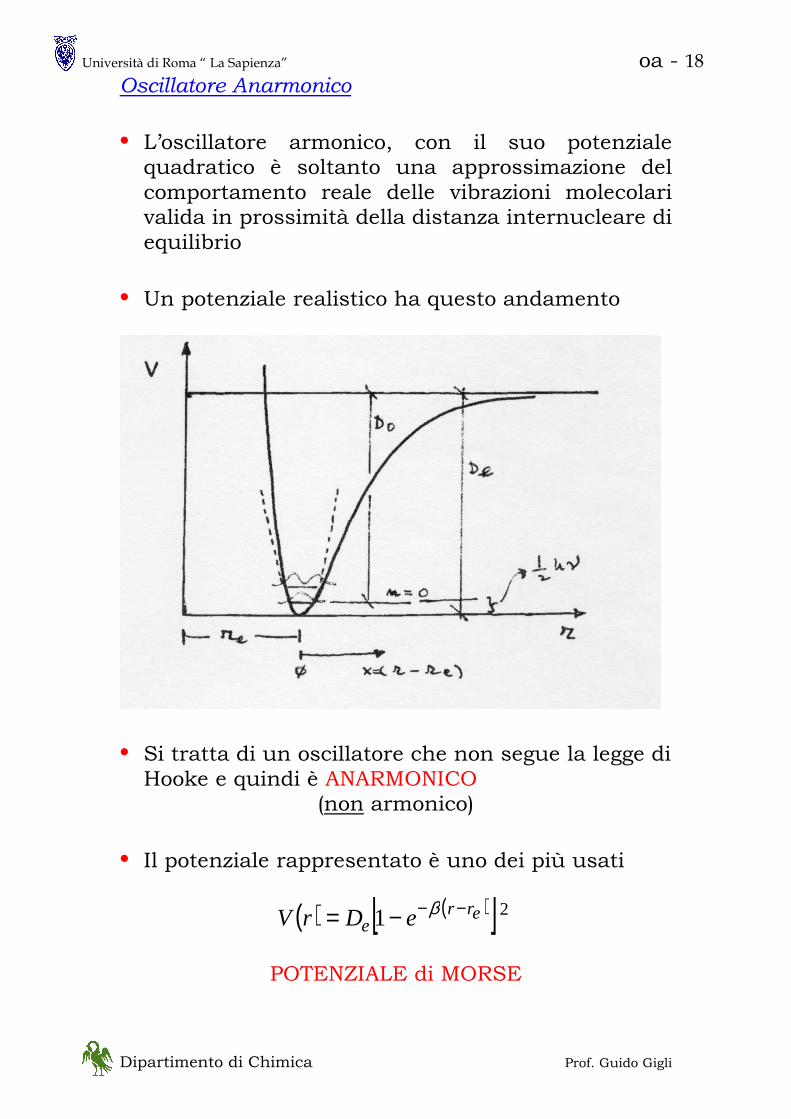
Oscillatore Anarmonico
• L’oscillatore armonico, con il suo potenziale quadratico è soltanto una approssimazione del comportamento reale delle vibrazioni molecolari valida in prossimità della distanza internucleare di equilibrio
• Un potenziale realistico ha questo andamento
• Si tratta di un oscillatore che non segue la legge di Hooke e quindi è ANARMONICO
(non armonico)
• Il potenziale rappresentato è uno dei più usati
( ) ( )[ ]21 erre eDrV −−−= β
POTENZIALE di MORSE
Università di Roma “ La Sapienza” oa - 19
Dipartimento di Chimica Prof. Guido Gigli
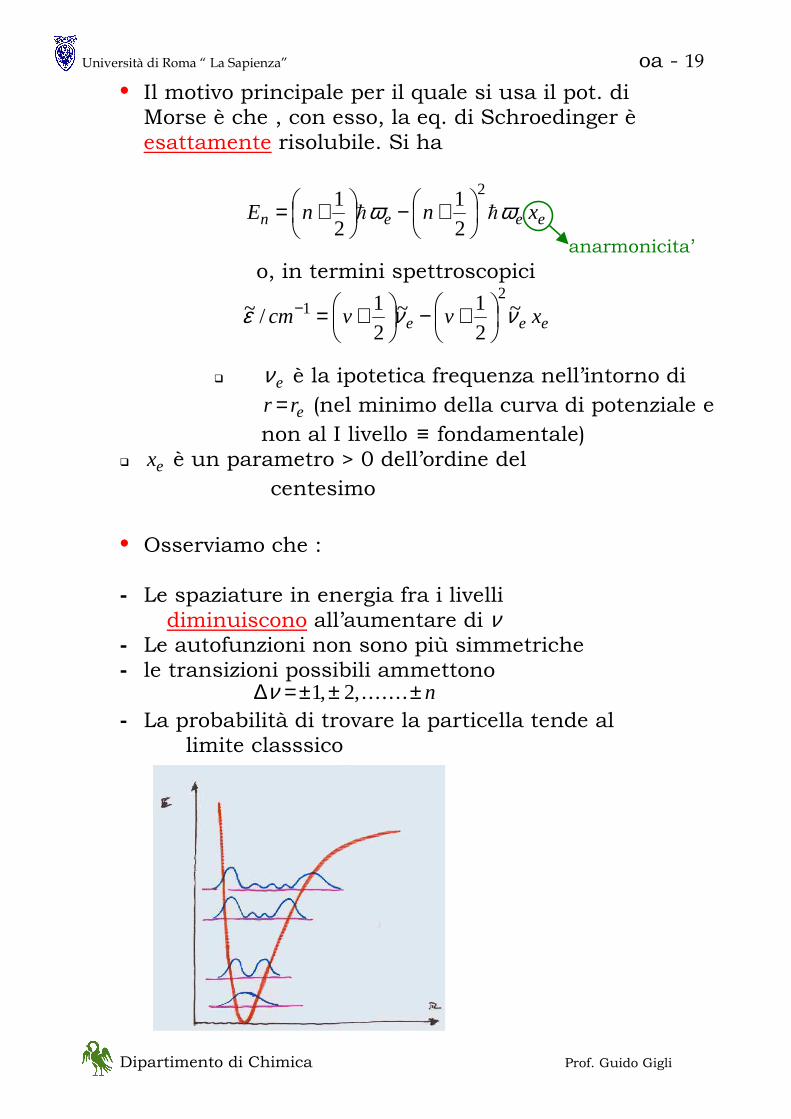
• Il motivo principale per il quale si usa il pot. di Morse è che , con esso, la eq. di Schroedinger è esattamente risolubile. Si ha
eeen xnnE ωω hh
2
21
21
+−
+=
o, in termini spettroscopici
eee xvvcm ννε ~21~
21
/~2
1
+−
+=−
� eν è la ipotetica frequenza nell’intorno di err = (nel minimo della curva di potenziale e
non al I livello ≡ fondamentale) � ex è un parametro > 0 dell’ordine del
centesimo
• Osserviamo che : - Le spaziature in energia fra i livelli
diminuiscono all’aumentare di ν - Le autofunzioni non sono più simmetriche - le transizioni possibili ammettono
n±±±=∆ .......,2,1ν
- La probabilità di trovare la particella tende al limite classsico
anarmonicita’
Università di Roma “ La Sapienza” rr - 1
Dipartimento di Chimica Prof. Guido Gigli
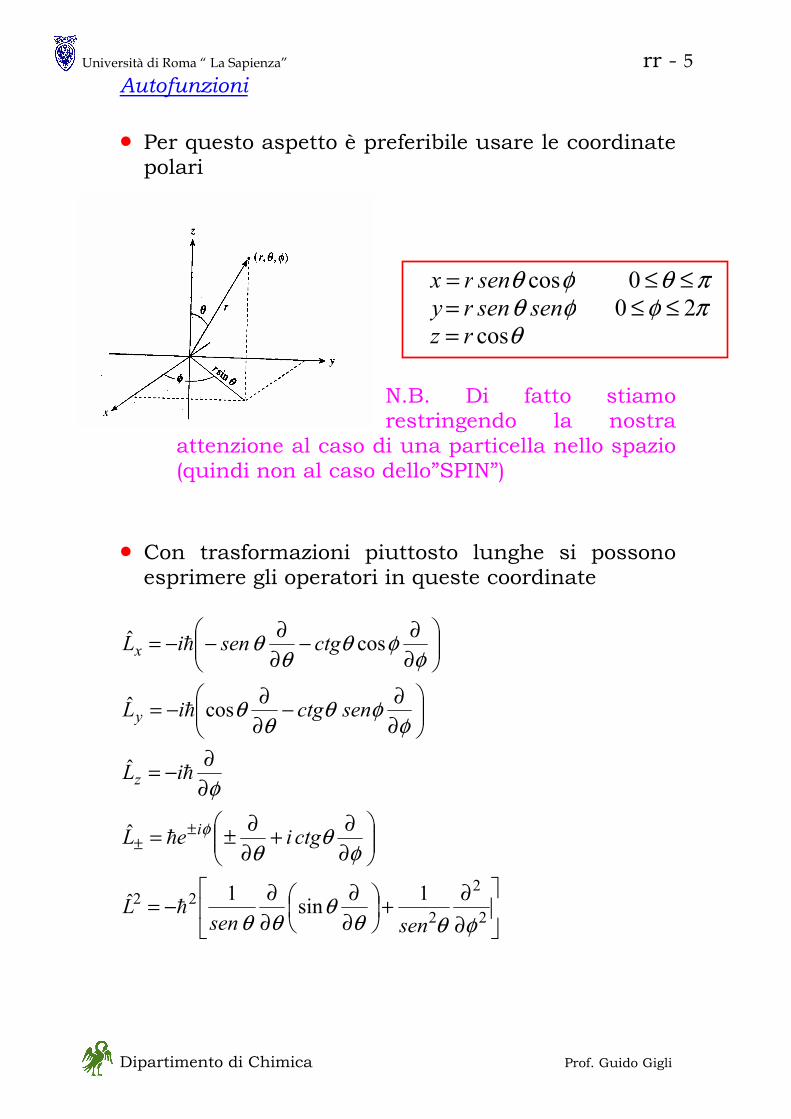
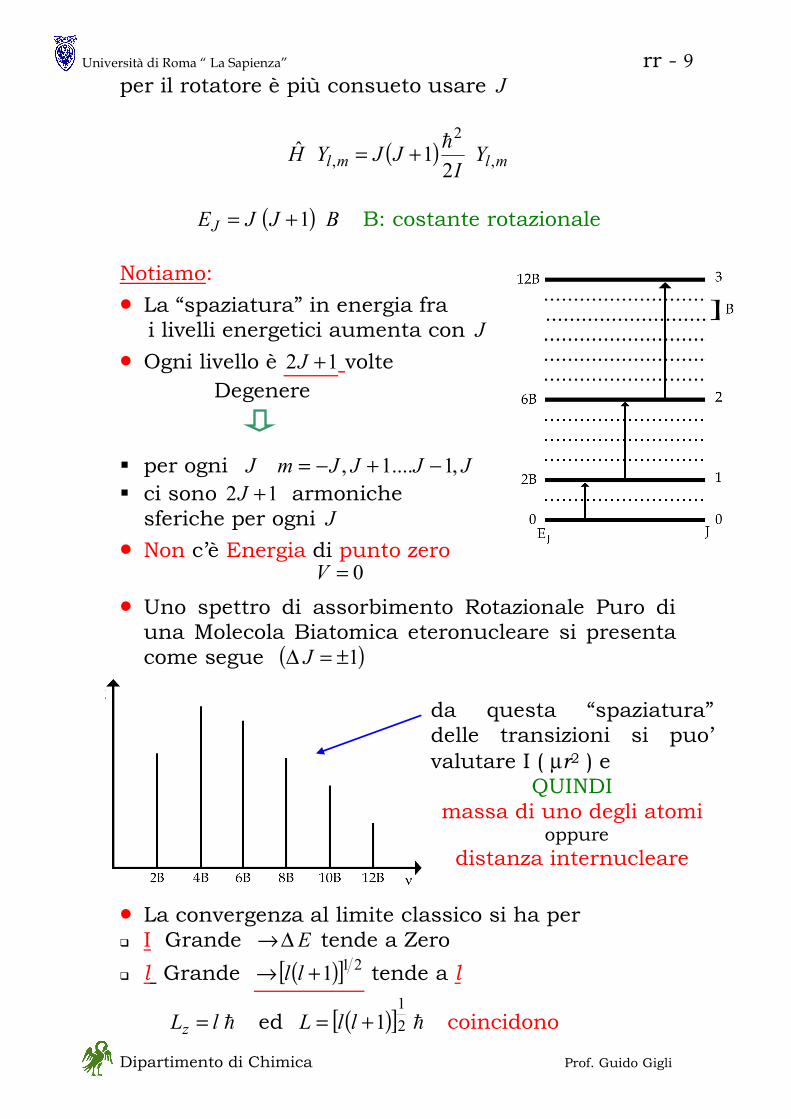
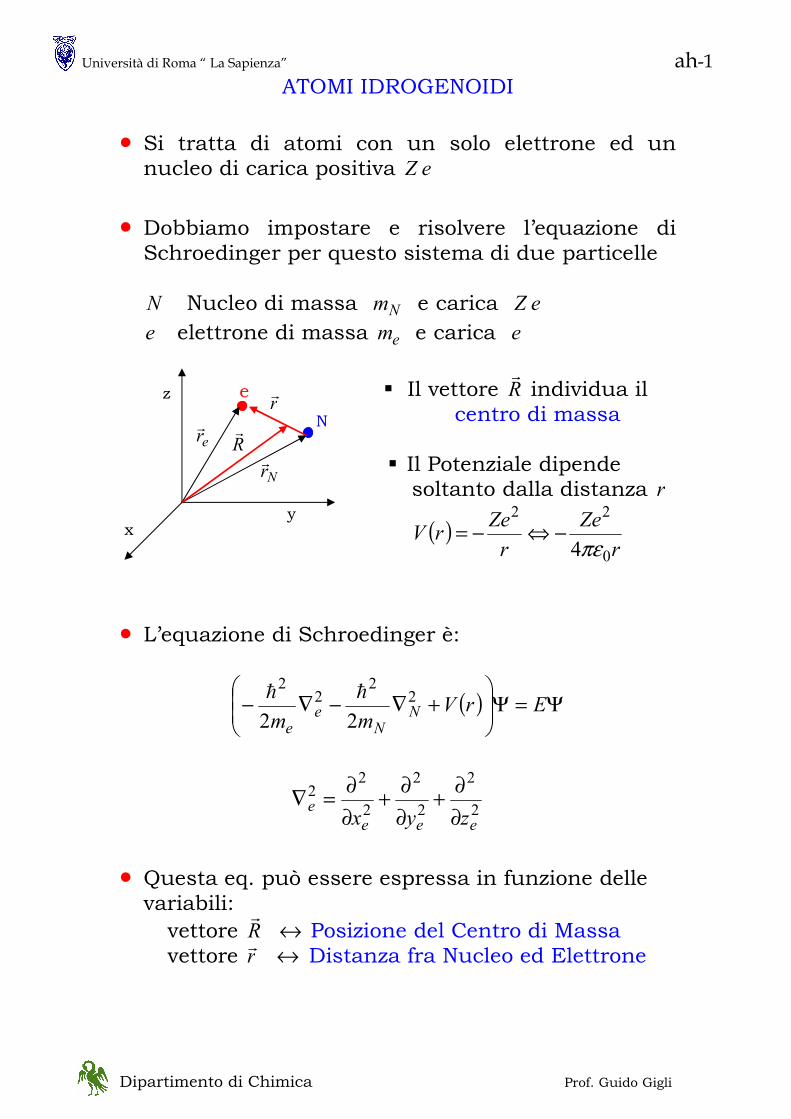
ROTATORE RIGIDO
Rotatore Rigido Classico
costante== 0V
tdd
rv
rv θθα
ππω ===
== &&2
2
222
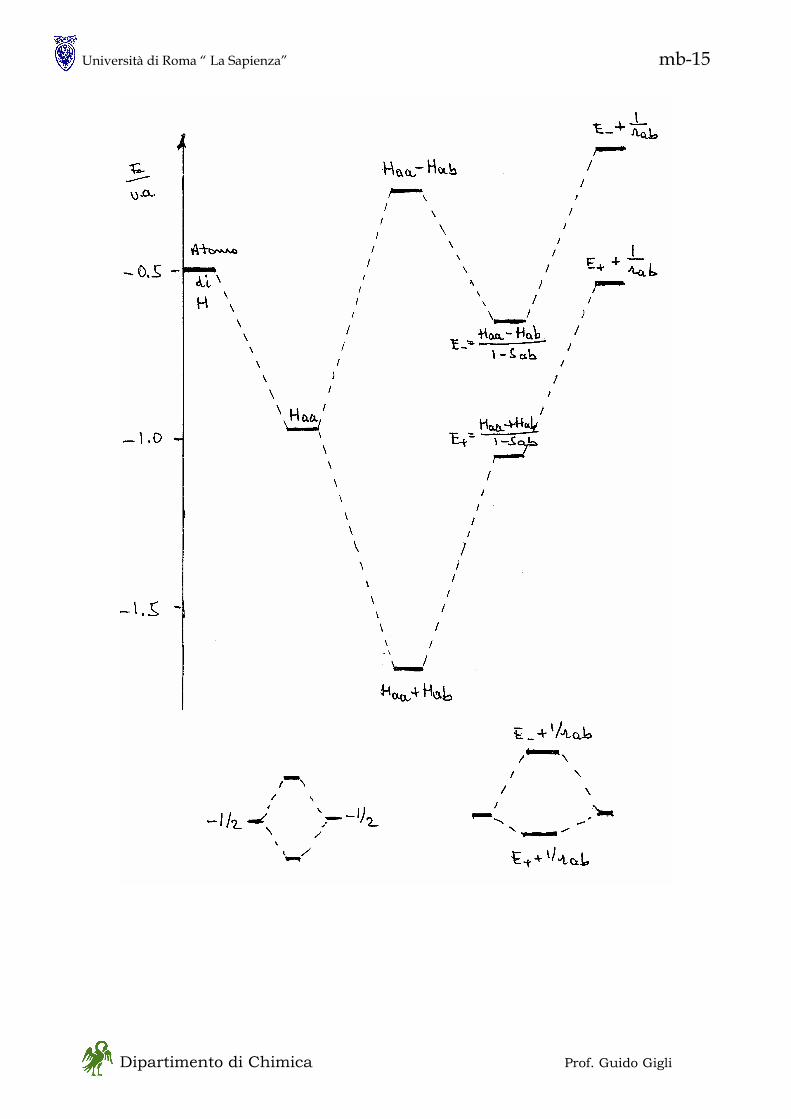
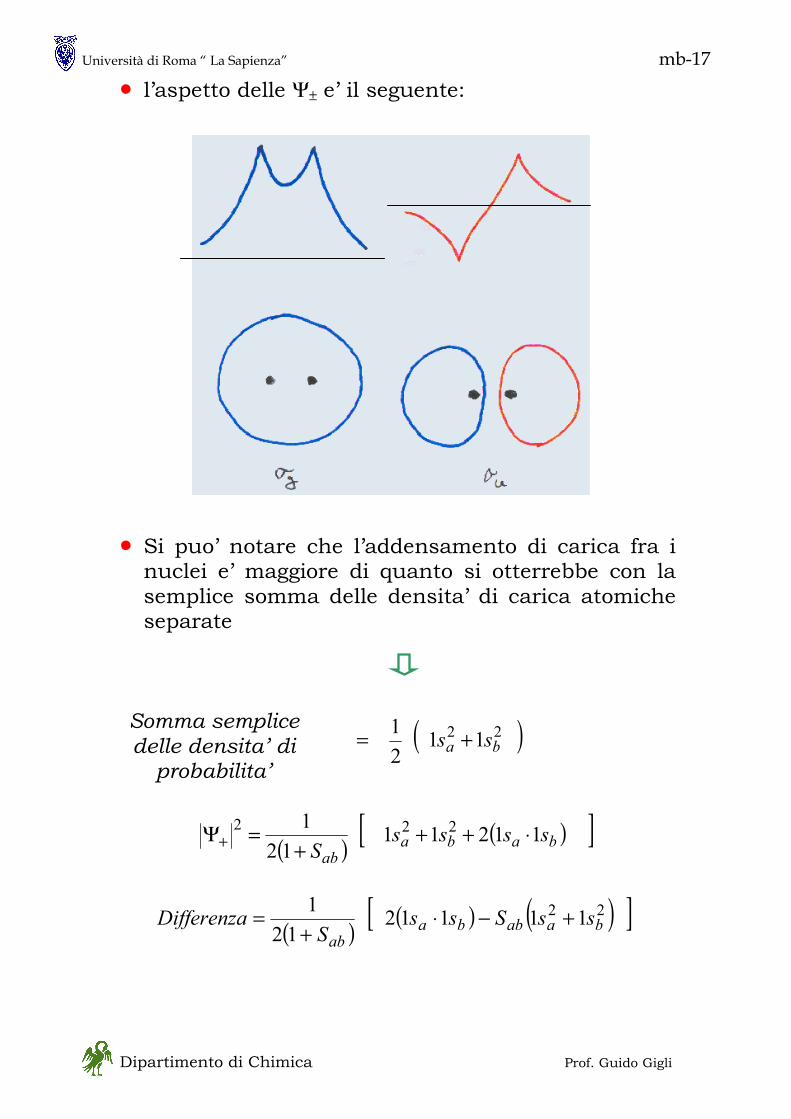
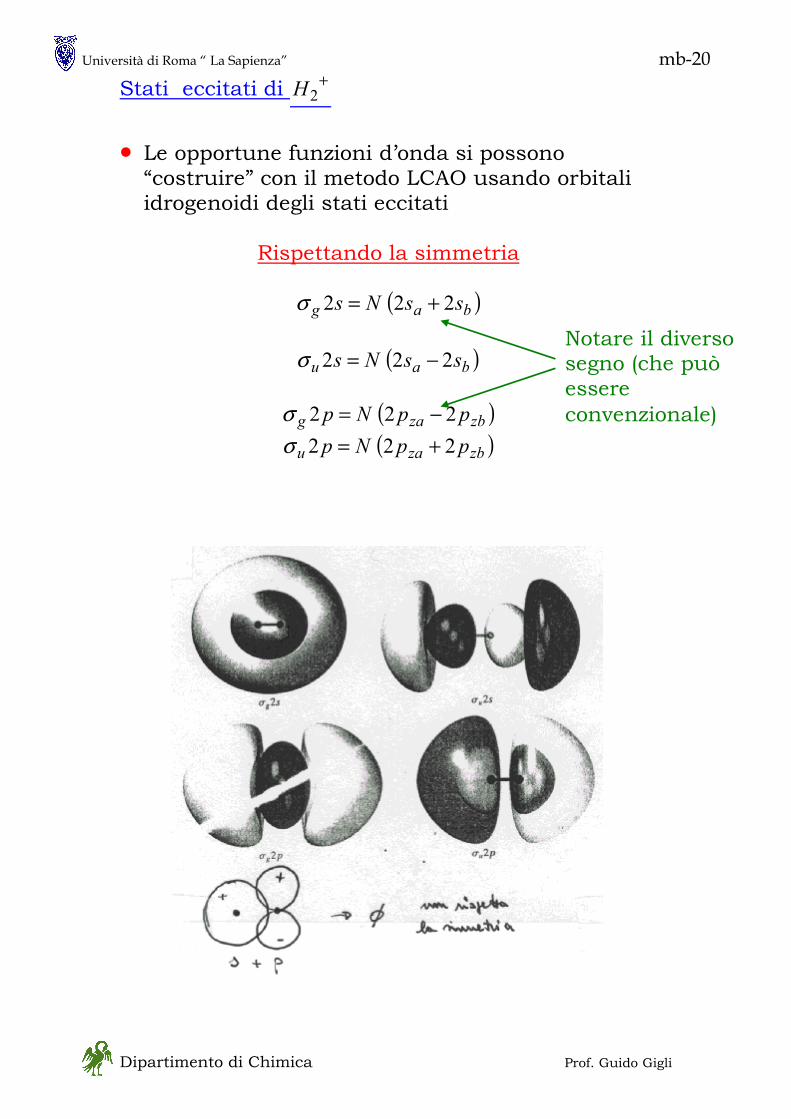
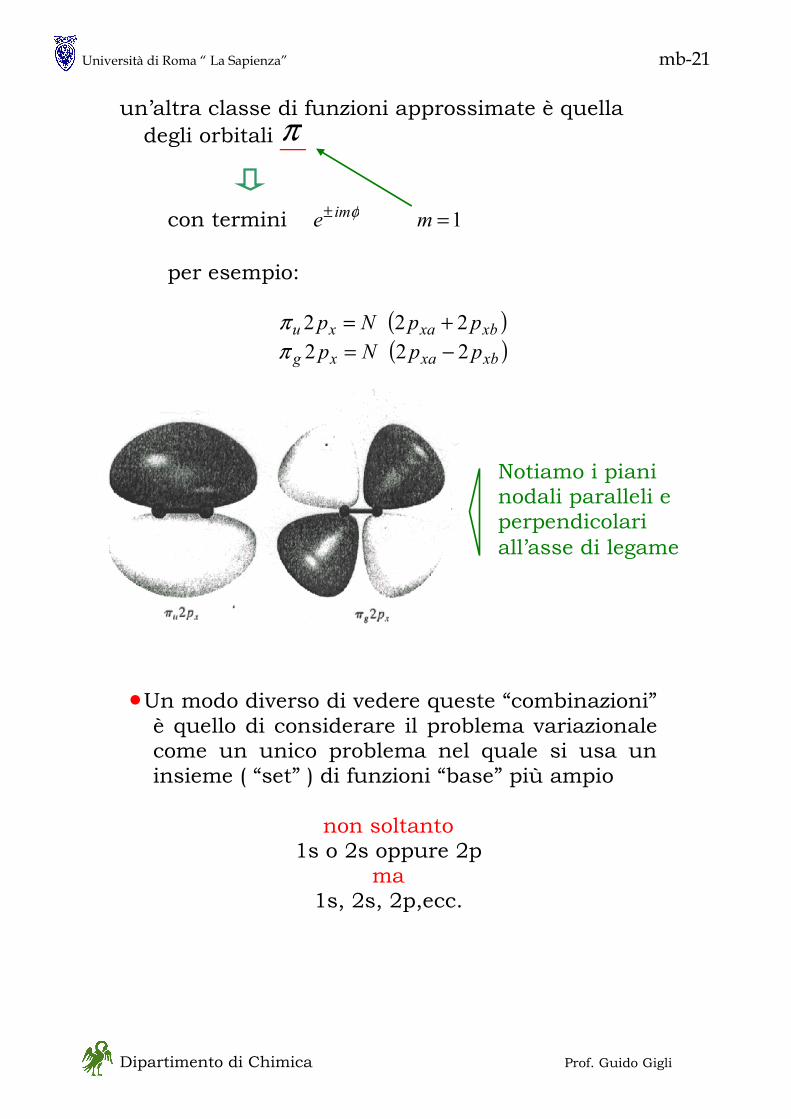
1
2
22
1
2222
211
21
21
21
21
21
ωω
ω
Irm
rmvmvmT
ii
i
ii
ii
==
=+=
∑
∑
=
=
momento di inerzia
rmm
mrmm
mrmrm
21
1
21
2
2211
+=
+=
+==
21
21
r e r
rrr e
( ) ( )( )
( )2
22
21
212122
21
21
22
221
22
1
r
rmm
mmmmrmm
mmrmm
mmI
µ=
++=
++
+=
2222221
21
21 ααµωµ && IrrT ===
• Introducendo il momento angolare
ααω && IrmrmvmrprLM ====×== 22
2222
21
21
2αωµ &Ir
ILT ===
r1
r2
Per passare agli aspetti quantistici dobbiamo occuparci del MOMENTO ANGOLARE dal punto di vista quantistico
Anche qui il moto di due masse vincolate è riconducibile a quello di una unica massa
fittizia µ sulla superficie di una sfera di raggio r
Università di Roma “ La Sapienza” rr - 2
Dipartimento di Chimica Prof. Guido Gigli
Momento Angolare • Abbiamo già visto che [ ][ ][ ]
ecc. dove
∂∂−
∂∂−=
=
=
=
yz
zyiL
LiLL
LiLL
LiLL
x
yxz
xzy
zyx
h
h
h
h
ˆ
ˆˆ,ˆ
ˆˆ,ˆ
ˆˆ,ˆ
e che zyx LLLL 2222 ˆˆˆˆ ++= commuta con le componenti
[ ] [ ] [ ]zyx LLLLLL ˆ,ˆˆ,ˆ0ˆ,ˆ 222 === • Abbiamo visto, quindi, che possiamo avere autofunzioni
simultanee di 2L ed una delle componenti zyx LLL ˆ,ˆ,ˆ oppureoppure
• Peraltro 2ˆ,ˆ,ˆ,ˆ LLLL zyx
sono Hermitiani
Deve esistere una Ψ per la quale
Ψ=Ψ
Ψ=Ψ
bL
aL
zˆ
ˆ2
ba , sono autovalori generici con dimensioni tE ⋅ ed ( ) 2tE
QUINDI
mmmL
mmL
z λλ
λλλ
h
h
=
=ˆ
ˆ 22
autovalori reali
Università di Roma “ La Sapienza” rr - 3
Dipartimento di Chimica Prof. Guido Gigli
• Se consideriamo, ora, 2L ed 2ˆzL i corrispondenti valori di aspettazione sono
222222
2222
ˆˆ
ˆˆ
hh
hh
mmmmmLmL
mmmLmL
zz ===
===
λλλλ
λλλλλλ
ma
2222 ˆˆˆˆ zyx LLLL ++=
QUINDI
22 ˆˆ zLL ≥
02 ≥≥ mλ
• Per trovare questi autovalori e le autofunzioni si puo’ usare il metodo, che non vedremo, degli operatori di Salita e Discesa
yx
yx
LiLL
LiLLˆˆˆ
ˆˆˆ
−=
+=
−
+
Autovalori
( )mlmmlL
mlllmlL
z h
h
=
+=ˆ
1ˆ 22
• l ed m sono, ovviamente, due numeri quantici • Ad ogni valore di l corrispondono 12 +l valori di
m
commutano con 2L ,ma non con zyxL ,,ˆ o fra di loro
l = 0, 1/2, 1, 3/2,.. m = -l, -l+1, …..l - 1, l
Università di Roma “ La Sapienza” rr - 4
Dipartimento di Chimica Prof. Guido Gigli
gli autovalori di 2L sono
( )12 +l volte degeneri • Il tutto può essere rappresentato così
• L’autovalore hm è la
proiezione zL del momento Angolare Totale
( )[ ] h
r 211+= llL • non tutte le orientazioni
di Lr
sono possibili
si ha la QUANTIZZAZIONE SPAZIALE
( )[ ] 211cos
cos
+=
=
llm
mL
θ
θ
hr
• con gli operatori di salita e discesa si “scala” la sequenza dei valori di m
Università di Roma “ La Sapienza” rr - 5
Dipartimento di Chimica Prof. Guido Gigli
Autofunzioni • Per questo aspetto è preferibile usare le coordinate
polari
N.B. Di fatto stiamo restringendo la nostra
attenzione al caso di una particella nello spazio (quindi non al caso dello”SPIN”)
• Con trasformazioni piuttosto lunghe si possono
esprimere gli operatori in queste coordinate
∂∂+
∂∂
∂∂−=
∂∂+
∂∂±=
∂∂−=
∂∂−
∂∂−=
∂∂−
∂∂−−=
±±
2
2
222 1sin1ˆ
ˆ
ˆ
cosˆ
cosˆ
φθθθ
θθ
φθ
θ
φ
φφθ
θθ
φφθ
θθ
φ
sensenL
ctgieL
iL
senctgiL
ctgseniL
i
z
y
x
h
h
h
h
h
φθ cossenrx = πθ ≤≤0φθ sensenry = πφ 20 ≤≤
θcosrz =
Università di Roma “ La Sapienza” rr - 6
Dipartimento di Chimica Prof. Guido Gigli
Notiamo che la variabile r non compare e che, quindi, le autofunzioni dipenderanno dalle variabili θ e φ
( )φθ ,, ,mlYml =
queste autofunzioni si chiamano ARMONICHE SFERICHE
e sono il prodotto di due funzioni in θ e φ ( ) ( ) ( )φθφθ mmlml FTY ⋅= ,, ,
notare che sono legate dal numero quantico m
• Il loro nome ha origine dal fatto che esse descrivono il comportamento di una sorta di “tamburo” sferico (un pianeta completamente allegato, un solido comprimibile)
• Si puo’ vedere che considerando la parte in φ la
funzione ( ) φφ imm eF = Deve essere a singolo valore
e continua ( )πφφ 2+= imim ee ( 12 =πime )
K,3,2,1,0 ±±±=m
Una semplice interpretazione fisica è che la funzione d’onda deve richiudersi su se stessa esattamente in un giro (notare l’analogia con la particella nella scatola per la quale la funzione d’onda deve andare a zero ai bordi)
ora m non puòassumere valori
seminteri
Università di Roma “ La Sapienza” rr - 7
Dipartimento di Chimica Prof. Guido Gigli
• Le espressioni analitiche complete sono assai
complicate
( ) ( ) ( )( )( ) ( )
( )( )
φ
πφ
θθ
θθ
imm
lml
mlm
l
ml
ml
eF
dd
mlmll
lT
21
221
,
21
sincos
sin!2
!12!2
1
=
+
−+−= +
++
• In forma più compatta usando i “polinomi (o
funzioni) associati di Legendre”
( )
( ) ( ) ( )
( ) ( )
) xdove (
θ
θ φ
cos
1!2
1
1
cos
2
22
,
=
−=
−=
=
l
ll
ll
lm
mmm
l
immlml
xdxd
lxP
xPxd
dxxP
ePNY
IN CONCLUSIONE
polinomi associati di Legendre
polinomi di Legendre
Università di Roma “ La Sapienza” rr - 8
Dipartimento di Chimica Prof. Guido Gigli
• Si puo’ notare che la forma analitica è del tipo:
• torniamo ora al problema del Rotatore Rigido Rotatore Rigido Quantistico
• Essendo la energia classica I
LE2
2= ne consegue
che dobbiamo cercare le soluzioni della eq. di Schroedinger ind. dal tempo ( )tfV ≠= ,0 per l’operatore H
ILHLH2ˆˆˆˆ2
2 =⇒∝
H ed 2L commutano ed hanno in comune
autofunzioni ed autovalori Autofunzioni
mlY , Armoniche Sferiche Autovalori
( ) mlmlml YI
llYLI
YH ,
2
,2
, 21ˆ
21ˆ h+==
Università di Roma “ La Sapienza” rr - 9
Dipartimento di Chimica Prof. Guido Gigli
per il rotatore è più consueto usare J
( ) mlml YI
JJYH ,
2
, 21ˆ h+=
( ) BJJEJ 1+= B: costante rotazionale
Notiamo: • La “spaziatura” in energia fra
i livelli energetici aumenta con J • Ogni livello è 12 +J volte
Degenere
per ogni JJJJmJ ,1....1, −+−= ci sono 12 +J armoniche
sferiche per ogni J • Non c’è Energia di punto zero
0=V • Uno spettro di assorbimento Rotazionale Puro di
una Molecola Biatomica eteronucleare si presenta come segue ( )1±=∆ J
da questa “spaziatura” delle transizioni si puo’ valutare I ( µr2 ) e
QUINDI massa di uno degli atomi
oppure distanza internucleare
• La convergenza al limite classico si ha per
I Grande E∆→ tende a Zero l Grande ( )[ ] 211+→ ll tende a l
hlLz = ed ( )[ ] h21
1+= llL coincidono
Università di Roma “ La Sapienza” ah-1
Dipartimento di Chimica Prof. Guido Gigli
ATOMI IDROGENOIDI
• Si tratta di atomi con un solo elettrone ed un nucleo di carica positiva eZ
• Dobbiamo impostare e risolvere l’equazione di
Schroedinger per questo sistema di due particelle
N Nucleo di massa Nm e carica eZ e elettrone di massa em e carica e
Il vettore Rr individua il
centro di massa
Il Potenziale dipende soltanto dalla distanza r
( )r
Zer
ZerV0
22
4πε−⇔−=
• L’equazione di Schroedinger è:
( ) Ψ=Ψ
+∇−∇− ErV
mm NN
ee
22
22
22hh
2
2
2
2
2
22
eeee zyx ∂
∂+∂∂+
∂∂=∇
• Questa eq. può essere espressa in funzione delle
variabili: vettore R
r ↔ Posizione del Centro di Massa
vettore ↔rr Distanza fra Nucleo ed Elettrone
Rr
rr
err
Nrr
e N
y x
z
Università di Roma “ La Sapienza” ah-2
Dipartimento di Chimica Prof. Guido Gigli
si ottiene, per via classica, un Hamiltoniano
( )rVM
H rR +∇−∇−= 22
22
22ˆ rr hh
µ che tiene conto di:
- energia cinetica dell’intera massa del sistema pari ad M = mN + me
- energia cinetica ed energia potenziale della particella fittizia di massa ridotta µ
Ne
Nemm
mm+
=µ
SE ( ) ( )tfrV ≠
( ) ( )rRErRH rrrr ,,ˆ Ψ=Ψ
Si separa facilmente
con ( ) ( ) ( )rRrR Ψ⋅=Ψ χ, ( R ed r sarebbero sempre vettori )
( ) ( )RERM rR χχ =∇− 22
2h
( ) ( ) ( )rErrV rr Ψ=Ψ
+∇− 2
2
2µh
IN SINTESI
Abbiamo ottenuto un risultato della massima importanza che, in generale,mostra che il moto traslazionale può essere separato ESATTAMENTE dai moti cosiddetti “interni”
Traslazione libera di una particella di massa M
Eq. di Schroedinger per una particella fittizia di massa µ che si muove in un potenziale V(r)
Università di Roma “ La Sapienza” ah-3
Dipartimento di Chimica Prof. Guido Gigli
• Per quanto riguarda l’energia
Interni MotieTraslazionTotale EEE += • Dal punto di vista fisico, e per quanto riguarda i
moti interni, tutto ciò equivale a considerare il nucleo pressochè fermo e l’elettrone in moto rispetto a questo
INFATTI
eee
e
eNeN
mmmm
mmmm
≈⋅=+
⋅≅≅
≅⇒>>
18011800
1180018001800
µµ poichè
a parità di tempo elettrone 1000 o
A
nucleo 0.1 o
A • Procediamo ora nella soluzione della eq. di
Schroedinger della particella µ nel potenziale ( ) ↔rV Potenziale Centrale
( )[ ] ( )[ ] ( )[ ] 0ˆ,ˆˆˆ,ˆ,ˆ 222222 =+⋅= LLrfLLrfLLrf a b a) i due operatori dipendono da variabili diverse b) un operatore commuta sempre con se stesso
♦ zL commuta con [ ] 0ˆ,ˆˆ 22 =zLLL
zL non dipende da r
QUINDI
[ ] 0ˆ,ˆ =zLH
Gli operatori zLedLH ˆˆ,ˆ 2 hanno autofunzioni in comune
Università di Roma “ La Sapienza” ah-5
Dipartimento di Chimica Prof. Guido Gigli
• Le autofunzioni di 2L ed zL (che non operano su r ) sono le armoniche sferiche ( )Φ,, θmlY
ogni funzione del tipo ( ) ( )Φ,θmlYrR
è ancora autofunzione
QUINDI
( ) ( )φθφθ ,,,,ˆ rErH Ψ=Ψ
( ) ( ) ( )( ) ( )φθφθ
φθφθ,,,,ˆ
,,1,,ˆ 22
rmrLrllrL
z Ψ=Ψ
Ψ+=Ψ
h
h
( ) ( ) ( )φθφθ ,,, ,mlYrRr =Ψ
che sostituita nella eq. di Schroedinger e dividendo per Ψ consente di scrivere
( ) ( ) ( ) ( ) ( ) ( )
( ) ( )( ) ( ) ( ) ErVll
rrRrRrT
EYrRrVLr
rTYrR ml
ml
=+++
=
++
22
,2
2,
12
1ˆ
,ˆ2
1ˆ,
1
hµ
φθµφθ
( ) ( ) ( ) ( )
e
eff
H
V
rRErRrVllrrd
drrd
dr
ˆ
ˆ
122 2
22
2
2=
+++
−
µµhh
Università di Roma “ La Sapienza” ah-6
Dipartimento di Chimica Prof. Guido Gigli
• Notiamo che:
H dipende dal numero quantico l
ci aspettiamo che anche le soluzioni ( )rR dipendano da l
Al potenziale centrale ( )rV che, nel nostro caso è
attrattivo, si somma un termine repulsivo che può essere interpretato come dovuto ad effetti centrifughi ( 0≠l )
Sino ad ora la trattazione è generale nel senso di essere applicabile a tutti i casi di due corpi interagenti ( p.e. V(r) potrebbe essere il potenziale di interazione fra due atomi in una molecola )
Per il caso dell’atomo Idrogenoide
( )r
ZerV2
−= mme =≅µ
• La soluzione della eq. ( ) ( )rRErRHe =ˆ è, come spesso accade, piuttosto difficile; ne vedremo soltanto i risultati ma prima di cio’ introduciamo delle unità di misura assai usate in chimica quantistica: le unità atomiche
• Si tratta di unità di misura che consentono di
ottenere espressioni analitiche piu’ semplici ed aver a che fare con numeri dell’ordine di grandezza dell’unità
Università di Roma “ La Sapienza” ah-7
Dipartimento di Chimica Prof. Guido Gigli
Lunghezza
2
2
0 ema h= ) se ( 1052918.0A52918.0 10
emm =⋅== − µo
BOHR ( raggio della I orbita dell’atomo di Bohr)
massa Kggaume
3128 1010.91010.9..1 −− ⋅=⋅==
tempo se
a 172
00 104189.2 −⋅== hτ
tempo impiegato da 1 elettrone 1s dell’atomo di H per percorrere 1 bohr carica Ce 1910602189,1 −⋅= carica dell’elettrone
energia ) 1s elettrone un diTOTEHartreeae (21
0
2⋅−==
= - 2 13.606 eV = 27.212 eV momento angolare h
Campo elettrico 20ae
QUINDI
Per esempio, con queste unità, l’Hamiltoniano dell’atomo idrogenoide ( con µ = µε ) diventa:
reZ
mH
e
22
2
2ˆ −∇−= h
rZH −∇−= 2
21ˆ
campo elettrico di un protone alla distanza di 1 bohr
Università di Roma “ La Sapienza” ah-8
Dipartimento di Chimica Prof. Guido Gigli
Autofunzioni
( )
⋅
⋅
−⋅= +
+0
12
00
22expanZrL
narZ
naZrNrR l
ln
l
nl
( 12 ++l
lnL sono funzioni complicate dette polinomi associati di Laguerre ed N è un fattore di normalizzazione) complessivamente, quindi, si ha:
( ) ( )φθ ,lmnlnlm YrR=Ψ
( )( )[ ]
( )φθ ,22exp
!2!12
,0
12
00
21
3
3
0
mll
ln
l
nlm
YanZrL
narZ
naZr
lnnln
anZ
⋅
⋅
⋅
−
⋅
+−−
−=Ψ
++
lnlm mlnmln ,,,, ==Ψ
per gli elettroni si usa lm • Come si vede dall’equazione differenziale che si era
ottenuta, per ogni valore del numero quantico l ci sono infinti valori di n che soddisfano tutte le condizioni fisiche volute; si dimostra che n puo’ assumere soltanto i valori l+1, l+2, ..ecc.
• Complessivamente i numeri quantici che compaiono nelle autofunzioni possono assumere soltanto alcuni valori e sono fra loro correlati
n = 1, 2, 3,... numero quantico principale l = 0, 1, 2…(n-1) “ “ orbitale od
azimutale lm = 0, ± 1, ± 2 l± “ “ magnetico
Università di Roma “ La Sapienza” ah-9
Dipartimento di Chimica Prof. Guido Gigli
• Sostituendo i vari valori di lmln ,, si ha:
( )
( )
( )
( )
( )φθπ
φθθπ
θπ
φθπ
θπ
π
φθπ
θπ
π
π
i2sena3rZ
arZ
aZ
1621
isena3rZ
arZ
aZ
811
13a3rZ
arZ
aZ
6811
isena3rZ
arZ
arZ6
aZ
811
a3rZ
arZ
arZ6
aZ
812
a3rZ
arZ2
arZ1827
aZ
3811
isena2rZ
arZ
aZ
81
a2rZ
arZ
aZ
241
a2rZ
arZ2
aZ
241
arZ
aZ1
2
00
2223
0223
00
2223
0123
2
00
2223
0023
000
23
0113
000
23
0013
02
0
22
0
23
0003
00
23
0112
00
23
0012
00
23
0002
0
23
0001
±
−
=Ψ
±
−
=Ψ
−
−
=Ψ
±
−
−
=Ψ
−
−
=Ψ
−
+−
=Ψ
±
−
=Ψ
−
=Ψ
−
−
=Ψ
−
=Ψ
±
±
±
±
expexp
expcosexp
cosexp
expexp
cosexp
exp
expexp
cosexp
exp
exp
,,
,,
,,
,,
,,
,,
,,
,,
,,
,,
• Da notare che la parte in r è del tipo
( ) ( ) )(expPolinomiocostante 1, n
rZrrrR lnlln −⋅⋅= −−
Università di Roma “ La Sapienza” ah-10
Dipartimento di Chimica Prof. Guido Gigli
nella funzione Rn,l(r) compare, quindi, un polinomio in r di grado n-1 con piu’ o meno termini a seconda del valore di l. • Le funzioni radiali sono normalizzate:
∫∞ =0
22 1drrR dove è importante notare che si è usata la parte in r dell’elemento di volume in coordinate polari:
drddr φθθsen2 Autovalori Gli autovalori dell’energia dipendono dal solo numero quantico n
2
2
0
2
2
2
22
42
222 nZE
ae
nZ
neZmE nn −=⋅−=−=h
Poichè l’energia dipende soltanto da “n ” ma vi sono vari valori di “l ” ed “m” per ogni valore di “n ” ogni stato del sistema è degenere ( )12...75312 −++++== nngn
Infatti: ( )
( )
( )
44444 844444 76
K
MMMM
K
∗
+−
−
+= ∗
∗
n
11n27531
1n3210
1l2m
l
oppure:
( ) ( )∑∑−
=
−
==+−=+
−=+=+
1
0
221
0 212212
n
l
n
lnnnnnnnnll
La notazione è la seguente l 0 1 2 3 simbolo s p d f sharp principal diffuse fundamental
Università di Roma “ La Sapienza” ah-11
Dipartimento di Chimica Prof. Guido Gigli
• Vediamo l’andamento della parte radiale delle autofunzioni • E’ utile analizzare la probabilità di trovare
l’elettrone ad una distanza r dal nucleo; ossia in una corteccia sferica di spessore dr
( ) ( )∫ ∫ =ΨΨ
=Ψπ π φθφθφθ0
20
*
2
,,,,
angoli gli tutti su integrata
ddrdrr nlmnlm
=∫ ∫ φθθπ π ddYYdrrrR lmlmnl sen)( 020
*22
1)( 22 ⋅drrrRnl ( le armoniche sferiche sono normalizzate )
• si ha, quindi, la funzione di distribuzione radiale
( ) drrRdrrP 22=
Il numero di “nodi” e’ pari ad n -l -1
A parita’ di n la probabilita’ di trovare un elettrone vicino al nucleo diminuisce con l
Università di Roma “ La Sapienza” ah-12
Dipartimento di Chimica Prof. Guido Gigli
• da notare che per l’orbitale 1s la distanza a cui si ha massima probabilita’ di trovare l’elettrone (rprob) e’ proprio in corrispondenza del raggio di Bohr
• è anche istruttivo valutare la distanza media dell’elettrone dal nucleo
==ΨΨ= ),()(),()( φθφθ lmnllmnlnlmnlm YrRrYrRrr
==
=
∫∫ ∫∫
∫ ∫ ∫∞∞
∞
032
020
*0
32
20 0
20
*
sen
sen
drrRddYYdrrR
dddrrYRrYR
φθθ
φθθπ π
π π
+− 2
2
0 2)1(
23
nll
Zna
dove si puo’ notare che - per Z=1, n=1, l=0 ( cioè per lo 1s di H ) rmedio = 1.5
a0
- all’umentare di Z diminuisce rmedio - rmedio(2s) ≠ rmedio(2p) anche se i due orbitali sono
degeneri ( come mai?) • Consideriamo ora anche la parte angolare
per 0=l la simmetria è sferica sia per Ψ che 2Ψ
rprob rmedio
Università di Roma “ La Sapienza” ah-13
Dipartimento di Chimica Prof. Guido Gigli
per 0≠l si devono considerare le forme delle armoniche sferiche
e della “deformazione” delle stesse dovuta alla parte radiale
vediamo il solo caso degli ORBITALI p
θcos2
exp0
2020,1,2 ⋅
−⋅⋅=Ψ=Ψ=Ψ
arZrpzp cost
per la sola
parte angolare
termine dovuto a Mulliken che ricorda le orbite dell’atomo di Bohr
Università di Roma “ La Sapienza” ah-14
Dipartimento di Chimica Prof. Guido Gigli
• Comunque quella che interessa di più è la intera Ψ che si rappresenta bene con “curve di livello” le quali uniscono i punti nei quali la funzione assume il medesimo valore o modulo 2Ψ
12 ±Ψ p • Si tratta di funzioni immaginarie
φθ iesenarZr ±
−⋅⋅
02expcos t
Peraltro
12*
1212*
12 +−−+ Ψ=ΨΨ=Ψ pppp
121212*
12 +−++ ΨΨ=ΨΨ pppp =
121212*
12 −+−− ΨΨ=ΨΨ pppp
i due orbitali 12 ±Ψ p hanno la medesima funzione densita’ di probabilità
Università di Roma “ La Sapienza” ah-15
Dipartimento di Chimica Prof. Guido Gigli
INOLTRE
1212 −+ ΨΨ pp e anche una loro combinazione sono degeneri lineare è autofunzione di
H con lo stesso autovalore per rappresentarle usiamo una combinazione lineare che sia reale
( ) ( ) ( )
( ) Φ=
=+=Ψ+Ψ
±
−±−+
cos2sen2
1rR
eesen2
1rR2
1
1p2
ii1p21p21p2
θ
θ φφ
Φ=
Φ
−⋅=Ψ
cos
cos2
exp0
2
θ
θ
senrx
senarZrpx cost
Analogamente
( )
Φ=
Φ
−=Ψ=Ψ−Ψ −+
sensenry
sensena2rZrcost
2i1
0py21p21p2
θ
θexp
• Analoghe combinazioni lineari di autofunzioni
degeneri con termini immaginari le rendono reali (orbitali d, f, ecc )
• Possiamo, peraltro, notare che queste nuove
funzioni d’onda non rappresentano più autostati di
zL (anche se sono ancora autostati di H ed 2L )
Università di Roma “ La Sapienza” ah-16
Dipartimento di Chimica Prof. Guido Gigli
22
42
2 neZmEn
h−= per l’Idrogeno ( ) 22
4
2:
nemEn
h−=⇒=1Z
quindi per l’atomo di Idrogeno:
223
41
4 nR
ncmecmEn
∞− −=−=hπ
HHNHe
He
Ne
Rmmmm
mmRmm
=+
=
∞== ∞
µ
µ
se
se
Vi è un addensamento di livelli all’aumentare del numero quantico “n”
E’ interessante osservare l’andamento dei livelli per vari tipi di potenziale
Si nota la competizione fra due effetti
Aumento dell’intervallo fra i livelli all’aumentare dell’Energia
Diminuzione della spaziatura fra i livelli all’aumentare delle dimensioni spaziali nelle quali è confinata la particella
Costante di Rydberg= 109737 cm-1
Università di Roma “ La Sapienza” ah-17
Dipartimento di Chimica Prof. Guido Gigli
• Per avere una ulteriore idea della scala delle energie permesse rispetto al potenziale si può sovrapporre all’andamento del potenziale sresso i livelli della espressione 2nRE −=
• Ogni livello è, come già detto, degenere ( 2n ) così
come indicato nella ordinata di destra
Università di Roma “ La Sapienza” ah-18
Dipartimento di Chimica Prof. Guido Gigli
• Per rappresentare questa degenerazione ( per quanto riguarda il numero quantico “l” ) si possono rappresentare i livelli degeneri in posizioni adiacenti:
da notare che è un valore diverso dalla 1109737 −
∞ = cmR • E’ importante osservare che esistono anche
soluzioni al nostro Hamiltoniano, ( complesse da trovare ), con
E > 0
che non sono quantizzate Le autofunzioni corrispondenti sono fondamentali nello studio di Collisione elettrone ione fenomeni di “scattering”
con le opportune Regole di Selezione si possono indicare le transizioni in un Diagramma di Grotrian
1,01
±=∆±=∆
=∆
lmln qualsiasi
Stati non legaticontinuo di Energia
(oltre la Ionizzazione )
Università di Roma “ La Sapienza” ah-19
Dipartimento di Chimica Prof. Guido Gigli
• Terminiamo la trattazione con due osservazioni su:
Caratteristiche degli orbitali
Tutti gli orbitali con 0=l hanno valore finito sul nucleo “ “ 0≠l “ “ nullo “ “ la forma dello s1 può essere considerata un
compromesso fra - Bassa cinE piccola curvatura di Ψ - Bassa V grande energia cinetica Negli atomi Idrogenoidi all’aumentare di Z la distanza
di massima probabilità diminuisce
La carica più grande attira verso il nucleo l’elettrone
Energia degli orbitali • L’energia dipende dal numero atomico Z
QUINDI
E’ sempre più in basso in energia ( aumenta in modulo) all’aumentare di Z
Ψ V
r
per lo He+ è quattro volte piu’ grande
Università di Roma “ La Sapienza” ah-20
Dipartimento di Chimica Prof. Guido Gigli
Ulteriori Complicazioni
• Abbiamo visto che siamo in grado di spiegare lo spettro di un atomo di Idrogeno
Se si applica un campo magnetico la degenerazione degli stati di moto con il medesimo l e diverso m viene rimossa
Questo lo possiamo spiegare perchè all’elettrone in moto è associato un momento magnetico che interagisce con il campo magnetico esterno
• Se, però,si usa un campo magnetico molto forte e
si osserva lo spettro con grande risoluzione non si riesce a spiegare la comparsa di 6 transizioni che generano 5 righe spettrali
• Un altro esperimento di natura completamente diversa, impone di considerare qualcosa d’altro
STERN GERLACH (già visto nei lucidi “in”)
Mettiamo in evidenza la quantizzazione spaziale
Ci aspettiamo 3
transizioni
struttura FINE
Università di Roma “ La Sapienza” ah-21
Dipartimento di Chimica Prof. Guido Gigli
• Il nuovo concetto che deve essere introdotto per spiegare la struttura fine è lo SPIN
E’ cosa del tutto nuova e non vi sono analogie classiche
Di conseguenza non si può procedere nel modo
usuale: Esprimere la grandezza classica in funzione di momenti e coordinate
Sostituire questi momenti e coordinate con gli operatori appropriati
Si deve procedere introducendo altri postulati
quantomeccanici ( in realtà nella formulazione relativistica di DIRAC lo SPIN compare naturalmente)
I ) Gli operatori del momento angolare di Spin
commutano e si combinano come gli operatori del momento angolare normale
ecc
ecc
zxyyx
zz
SiSSSS
SSSS
ˆˆˆˆˆ
0ˆˆˆˆ 22
h=−
=−
Università di Roma “ La Sapienza” ah-22
Dipartimento di Chimica Prof. Guido Gigli
II ) Vi sono soltanto due autofunzioni ( α e β ) degli operatori di momento angolare di spin. Gli autovalori corrispondenti sono:
ββ
αα22
22
)1(ˆ)1(ˆ
h
h
+=
+=
ssS
ssS 21=s
hr
43=S
βββαααhh
hh
21ˆ21ˆ
−==
==
sz
sz
mS
mS 21±=sm
per la notazione: sms, e ⇒βα
↓=−=↑== 21,2121,21 βα
N.B. La parte difficile è che queste funzioni α e β sono funzioni del tutto astratte di uno spazio anch’esso astratto.
Si può generalizzare:
L’entità del momento angolare di spin di una particella è determinato dal numero quantico di spin
questo numero quantico ha un UNICO valore
positivo (intero o semintero ) che è caratteristico della particella (per esempio:
non23=s 2
3,1,21,0=s )
( anche ad ogni nucleo e’ associato uno spin caratteristico )
La distinzione fra particelle con spin intero ( BOSONI ) e particelle con spin semintero (FERMIONI ) è di tipo fondamentale Seguono forme diverse del principio di esclusione di PAULI
Università di Roma “ La Sapienza” ah-23
Dipartimento di Chimica Prof. Guido Gigli
• Da quanto detto si evince che l’orientazione del momento angolare di spin è quantizzata
III ) L’elettrone ( con spin) agisce come un piccolo
magnete il cui momento di dipolo è:
( )cm
ege
s 2−××= spin di angolare momentoµr
- Il fattore sg è postulato essere pari a due volte il
fattore classico. In realtà sg =2.0023227 ; valore previsto dalle teorie moderne di elettrodinamica quantistica
- elettronedell' icogiromagnet Rapporto →=− γcm
ege
s 2
Una finzione relativa allo spin è: Lo spin è il momento angolare che consegue alla rotazione di un corpo intorno al proprio asse
Le particelle cariche con spin possiedono un momento magnetico intrinseco
hr
23=S
Sz/h s =1/2
Università di Roma “ La Sapienza” ah-24
Dipartimento di Chimica Prof. Guido Gigli
• Da quanto si è visto emerge che l’elettrone
dell’atomo idrogenoide possiede sia un momento angolare orbitale che un momento angolare di spin
( ) ( ) h
rh
r11 +=+= ssslll
hh szlz msml == • I contributi di questi due momenti angolari elettronici si
combinano per fornire un momento angolare elettronico totale
slj rrr
+=
Per analogia con i casi precedenti:
( ) hr
1jjj +=
jjjmmj jjz ++−−== Kh ,1, • Vediamo i possibili valori di j
r per valori particolari
di sl rr di e
• Consideriamo le componenti di sl rr
di e
( )
hh
hhh
21,
21
,,.........1,
+−=
++−−=
z
z
s
llll
la componente zj sarà la somma delle componenti
zz sl ed considerando tutte le somme possibili
Università di Roma “ La Sapienza” ah-25
Dipartimento di Chimica Prof. Guido Gigli
• Per esempio :
con ( ) hh
hh
2121211011
+−==+−=→=
z
z
ssll
si ha per 23
21
21
21
21
23 −−−=→ hzz jj
lo stato 21
21 == jjz da derivah
ma per 21
21
21 −== jmj
lo stato 23
23 == jjz da derivah
ma per 23
21
21
23
23 −−== jmj
• Allo stesso risultato ( quando c’è un solo elettrone ) si perviene sommando e sottraendo sl ed
211 == sl
2123 =−==+= sljslj Si può generalizzare questa considerazione :
♦ Quando vi sono due momenti angolari ( con numeri quantici 21 jj e ) anche il momento angolare totale è quantizzato ( con modulo
( )[ ] h211+JJ ) ed il numero quantico J è fornito dai termini della cosiddetta serie di Clebsch-Gordan
( ) ( ) 212121 ,,1, jjjjjjJ −−++= K
• Nel nostro esempio un elettrone ( )1=lp dà luogo a due stati di moto con diverso momento angolare totale
per qualsiasi valore di di l ≠ 0 si hanno due valori di j
Università di Roma “ La Sapienza” ah-26
Dipartimento di Chimica Prof. Guido Gigli
• Questi stati con diverso momento elettronico angolare totale hanno energia diversa
• L’origine della differenza in energia è l’accoppiamento SPIN-ORBITA che nasce dalla interazione fra i momenti magnetici associati ai momenti angolari orbitali e di spin
per una descrizione quantitativamente corretta di queste interazioni è necessaria una trattazione quantomeccanica relativistica ( Dirac 1928 )
• Non è più sufficiente specificare con un simbolo la
configurazione ( .....2,2,1 pss ) e,quindi j
S L12 + LS 12 + specifica il termine dove per 3210=l J
S L12 + specifica il livello KFDPSL = • Ne consegue che per un atomo di Idrogeno si
hanno i livelli seguenti configurazione fndnpnsn→ livello
272
252
252
232
232
212
212 FFDDPPS→
ogni livello è 2J+1 volte degenere (ad ogni livello corrispondono 2J+1 stati ) • una notazione completa è, per esempio: 21
22 Pp
Università di Roma “ La Sapienza” ah-27
Dipartimento di Chimica Prof. Guido Gigli
• Il diagramma dei livelli energetici va, quindi, modificato inserendo lo splittamento dei livelli con diverso j
• Ci si deve attendere che per uno stesso termine Ls 12 + il livello con j più piccolo sia ad energia
minore perchè i momenti magnetici orbitale e di spin si accoppiano in modo attrattivo
zzzzzz sljslj +=−=
• Il risultato è il seguente diagramma di Grotrian: (da notare che lo “splitting” j diminuisce all’aumentare di ‘n’ e di ‘l’ )
Università di Roma “ La Sapienza” he-1
Dipartimento di Chimica Prof. Guido Gigli
ATOMI POLIELETTRONICI
ATOMO DI ELIO : Stato fondamentale • Impostiamone l’Hamiltoniano dei moti interni
trascurando la energia cinetica del nucleo ( assumiamo che, rispetto ad me, mN = ∞,)
12
2
2
2
1
222
221
2
22ˆ
re
reZ
reZ
mmH
ee+−−
∇−
∇−=
hh
T V T
in u.a.
( )( ) ( )
1221
1221
22
21
12ˆ1ˆ
121ˆ
rHH
rrZ
rZH
++=
+−−∇+∇−=
Hamiltoniani di un Atomo
Idrogenoide
non stiamo considerando lo SPIN
Università di Roma “ La Sapienza” he-2
Dipartimento di Chimica Prof. Guido Gigli
• La eq.di Schroedinger risultante
( ) ( )2121 ,,ˆ rrErrH rrrr Ψ=Ψ
8 non è separabile in eq. In 21 rr ed 8 non è risolvibile in forma chiusa
• Vediamone delle soluzioni approssimate Il modello a particelle indipendenti • Ignoriamo il termine che ne impedisce la
fattorizzazione: 12
1r
( ) ( )2211 ˆˆˆ rHrHH +=
usando il metodo della separazione
delle variabili la eq. si separa in due eq.
111ˆ Ψ=Ψ EH 222ˆ Ψ=Ψ EH
- ( ) ( ) ( )221121, rrrr rrrr Ψ⋅Ψ=Ψ
- 21 ˆˆ HH oppure sono Hamiltoniani dell’atomo Idrogenoide
- ( ) Ψ=Ψ 11 rr dell’atomo Idrogenoide =
= ( ) Hmln r 11,, Ψ=Ψ r
- eV E -4Hartees -4u.a. h 8.1084
24
24
22 22
2
21
2
−===−=
=−−=−−=n
Zn
ZE
Università di Roma “ La Sapienza” he-3
Dipartimento di Chimica Prof. Guido Gigli
• In realtà la Energia “vera” è hE904.2−
21 PIPIVe79.0eV54.424.6 =+
• Proviamo con Hamiltoniano completo con 12
1r
e la funzione d’onda prodotto di Ψ idrogenoidi HH
21 ΨΨ
eVEZZ
rZZ
HHE
h
HHHH
8.7475.285
122
ˆˆ
2
12
22
2121
−=−=+−=
=ΨΨ+−−=
=ΨΨΨΨ=ΨΨ=
• in pratica, poiché il termine 1/r12 fa parte dell’Hamiltoniano, si tiene conto della repulsione elettronica in modo “medio” anziché ignorarla del tutto
• inoltre, notiamo che usando una funzione di prova
veraprova EHE >ΨΨ= ˆ
Quello che abbiamo visto non è altro che un caso particolare del Metodo Variazionale
valore di aspettazione dell’energia
Università di Roma “ La Sapienza” he-4
Dipartimento di Chimica Prof. Guido Gigli
Metodo Variazionale • Il teorema Variazionale asserisce che:
Se Φ è una funzione a buon comportamento che soddisfa alle condizioni al contorno del problema di interesse il valore di aspettazione di H calcolato con la Φ soddisfa la disuguaglianza 0ˆ EHEprova ≥ΦΦ= dove 0E è la energia vera dello stato fondamentale
• Vediamo perchè:
0ˆ 00 ≥Φ−Φ==− EHIEEprova
espandiamo la Φ nelle autofunzioni di H
(per le quali iii EH Ψ=Ψˆ ) ∑ Ψ=Φ iic ΦΨ= iic
( )
( ) 0
ˆ
02
0*
0*
≥−=
−=Ψ−Ψ=
∑
∑∑
EEc
EEccEHccI
ii
ijiij jijiij ji δ
≥ 0 ≥ 0
Università di Roma “ La Sapienza” he-5
Dipartimento di Chimica Prof. Guido Gigli
• Qualunque sia la funzione di prova provaE non è mai minore di 0E
• Il metodo si presta ad essere usato con parametri
da ottimizzare minimizzando E
Si esegue la cosiddetta “ottimizzazione variazionale”
• per esempio con ( ) ( ) ( )21112,1 1121
Hs
Hsss Ψ⋅Ψ==Φ
dove ( ) rZHs eZ
arZ
aZ ⋅−=
−
=Ψ
*23*21
0
1*23
0
*
2111exp1
ππ
( Z✶ è un parametro )
provaEZZZ =
−− *
1652*2
−−==
∂∂
165*20
*ZZ
ZE
• VeEZZE h 49.778477.21627
165
1627 22
* =−=
−=
−−=
=
il termine con Z deriva dall’ Hamiltoniano mentre quelli con Z * hanno origine dalla funzione d’onda
Università di Roma “ La Sapienza” he-6
Dipartimento di Chimica Prof. Guido Gigli
• Notiamo che: ZZ <* • Si può andare oltre con funzioni di prova più
adattabili
♦ con due s1 idrogenoidi con cariche effettive diverse
18,219,1 *2
*1 == ZZ hprova E8757,2E −=
uno degli elettroni è più legato
la repulsione interelettonica viene ridotta consentendo alla funzione di prova di tener conto della CORRELAZIONE fra il moto degli elettroni
♦ Con funzioni a molti parametri si è ottenuto
903724375.2−=E con correzioni -Relativistiche -per il moto nucleare
( )1-
1-
cm 0.02
cm
±=
=
76.198310
69.198310
leSperimenta
HPI e
gli elettroni si schermano reciprocamente dalla attrazione del nucleo
Università di Roma “ La Sapienza” he-7
Dipartimento di Chimica Prof. Guido Gigli
Alcune osservazioni sul Metodo Variazionale • Le Energie convergono verso il valor vero più
rapidamente di quanto facciano le funzioni d’onda
- anche con funzioni d’onda scadenti si possono avere Energie buone
peraltro - A parte l’energia, altre grandezze ( per esempio il
momento di dipolo) possono essere scadenti come le Ψ
• La dimostrazione che abbiamo visto si riferisce alla
stima della energia del livello fondamentale 0E come limite inferiore della provaE
provaE limite superiore della veraE0 • Esistono altri metodi di tipo genericamente
variazionale che forniscono un limite inferiore • In casi particolari ( e con difficoltà ) il metodo
variazionale può essere esteso anche alla stima dei livelli eccitati
• Un importante caso particolare è quello delle
funzioni di prova che sono combinazioni lineari di un insieme non completo di adatte funzioni d’onda
♦ Sovrapposizione di stati ♦ variazionale lineare
lo vediamo in seguito
Università di Roma “ La Sapienza” he-8
Dipartimento di Chimica Prof. Guido Gigli
Un’alternativa per affrontare problemi non risolubili esattamente è la Teoria delle Perturbazioni • Ne vediamo la forma applicabile a sistemi non
dipendenti dal tempo • Supponiamo che per un qualche sistema si sia in
grado di risolvere la eq. di Schroedinger
0000ˆ iii EH Ψ=Ψ
Supponiamo che per un qualche sistema (che è quello di nostro interesse) non si sia capaci di risolvere la eq. di Schroedinger. ma l’Hamiltoniano di questo sistema sia simile ed esprimibile con
PHHH ˆˆˆ 0 +=
♦ Questa è la PERTURBAZIONE
♦ L’ipotesi è che questo termine
sia piccolo
autofunzioni noteed
autovalori noti
Università di Roma “ La Sapienza” he-9
Dipartimento di Chimica Prof. Guido Gigli
• Il nostro scopo è quello di mettere in relazione
autovalori autovalori e e autofunzioni autofunzioni del del Sistema non Sistema perturbato perturbato
Immaginiamo di “applicare” la perturbazione in modo graduale sì da avere una variazione continua dallo stato perturbato a quello non perturbato
matematicamente
PHHH ˆˆˆ 0 λ+= λλλλ 0 1
...33221 +++ HHH λλλ
♦ Se il tutto funziona le iΨ soluzioni di iii EH Ψ=Ψˆ
sono simili alle 0iΨ e le iE alle 0
iE ♦ Gli stati iΨ e 0
iΨ dovrebbero essere correlati nel senso che per ( )10 →λ il sistema descritto da 0
iΨ finisce in iΨ
Università di Roma “ La Sapienza” he-10
Dipartimento di Chimica Prof. Guido Gigli
• Si possono esprimere le autofunzioni iΨ e gli
autovalori iE in serie di potenze del parametro λ
2210 ˆˆˆˆ HHHH λλ ++=
2210
2210
iiii
iiii
EEEE λλ
λλ
++=
Ψ+Ψ+Ψ=Ψ
λ è una sorta di “etichetta” per tenere
conto del livello della approssimazione
λ non ha un vero significato fisico ed alla fine viene posto uguale ad 1
- Correzioni lineari in λ sono del I ordine “ quadratiche in λ sono del II ordine
(sperabilmente minori) • Procedendo nella elaborazione della teoria si
ottengono una serie di relazioni per i vari ordini di perturbazione che andranno usati per ricavare la Energia e la Funzione d’onda che ci interessa sommando i contributi
....
.....210
210
+Ψ+Ψ+Ψ=Ψ
+++=
iiii
iiii EEEE
Ordine Zero
0000ˆ iii EH Ψ=Ψ
non ci dice nulla di nuovo
è la eq. di Schroedinger per il sistema noto approssimazione di “ordine zero”
Università di Roma “ La Sapienza” he-11
Dipartimento di Chimica Prof. Guido Gigli
I Ordine – Autovalori
10101 ˆ iiiii HHE =ΨΨ=
• Questa relazione va cosi’ interpretata:
la correzione di energia al I ordine è il valore medio della perturbazione del I ordine sullo stato non perturbato
I Ordine - Autofunzioni
00
0101011
ˆ ticoefficien i dove
ji
ijji
ijjjii EE
Hcc
−
ΨΨ=Ψ=Ψ ∑
≠
e, quindi, complessivamente:
∑≠
Ψ−
ΨΨ=Ψ
ijj
ji
iji EE
H0
00
0101
ˆ
• La perturbazione modifica la funzione d’onda
sovrapponendo le altre funzioni d’onda di ordine zero
• L’entità di ogni coefficiente della sovrapposizione
dipende da:
1. “L’elemento di matrice” della perturbazione che “mescola” due stati
H1ji
Università di Roma “ La Sapienza” he-12
Dipartimento di Chimica Prof. Guido Gigli
2. La differenza di energia fra gli stati che vengono “mescolati”
Osservazioni ed Avvertenze • Dalla nostra esposizione sono esclusi i casi
degeneri
Il E∆ al denominatore va a zero
• Ovviamente vi sono espressioni utili per le correzioni di ordine superiore, per esempio
1100202 ˆˆ iiiii HHE ΨΨ+ΨΨ=
qui si può notare che si ha la E di II ordine con la
1Ψ del I ordine
in realtà con una Ψ corretta all’ordine n
si ha “ E “ “ “ 12 +n
Università di Roma “ La Sapienza” he-13
Dipartimento di Chimica Prof. Guido Gigli
L’atomo di Elio con la Teoria delle Perturbazioni • Avevamo l’Hamiltoniano
( ) ( )12
2112ˆ1ˆˆ
rHHH ++=
secondo la teoria delle perturbazioni
1010
201 ˆˆˆˆˆˆ HHHHHH +=++=
ORDINE ZERO • Separando le variabili 0
20
10 Ψ⋅Ψ=Ψ
00
10
101ˆ iEH Ψ=Ψ 0
202
02
02ˆ Ψ=Ψ EH
HEE 20 −= Z=2 hEE 40 −= II ORDINE • Le nostre 0
20
1 ΨΨ e sono gli s1 dell’Idrogeno
2132231230 111 ZrZrZrZr eeZeZeZ −−−− =⋅=Ψπππ
• Usiamo la espressione dell’Energia al I ordine
=ΨΨ=ΨΨ= 0
12
00101 1ˆr
HE
ovviamente come nelcaso a particelle indipendenti
Università di Roma “ La Sapienza” he-14
Dipartimento di Chimica Prof. Guido Gigli
hh
HHH
ZrZr
EE
EEEEEE
dddddrdrsenr
senrr
eeZ
75.22375.1852
85
1
10
212121222
12
112
220
12000
20
202
6
−=⋅−=
+−=+==
=⋅
⋅= −∞ −∞∫∫∫∫∫∫
φφθθθ
θπ
ππππ
• Riassumendo ecco alcuni esempi di calcoli
Università di Roma “ La Sapienza” he-15
Dipartimento di Chimica Prof. Guido Gigli
Spin nelle funzioni d’onda dell’atomo di Elio Sino ad ora abbiamo trattato l’atomo di Elio senza tener conto dello spin degli elettroni
Principio di PAULI Si tratta di un principio che riguarda la relazione fra
SPIN e SIMMETRIA delle Ψ
- Emerge naturalmente in una
trattazione Relativistica - Lo introdurremo come un
Postulato aggiuntivo • Particelle identiche sono classicamente DISTINGUIBILI
♦ Almeno in via di principio mediante la loro posizione (conoscendo la traiettoria)
quantisticamente INDISTINGUIBILI
♦ La traiettoria non ha più significato
Massa, Carica, eccIdentiche
Università di Roma “ La Sapienza” he-16
Dipartimento di Chimica Prof. Guido Gigli
♦ la 2Ψ ci fornisce la Densità
di probabilità ♦ Se in un dV troviamo una particella non possiamo sapere quale è
QUINDI
Per un sistema, per esempio, con due particelle la eq. di Schroedinger e le sue soluzioni devono essere identiche anche se scambiamo le particelle
( ) ( )21
22
221
2,
222,1ˆ qqV
mmH +∇−∇−= hh
( )2,1H è uguale ad ( ) simmetrico è ; HH ˆ1,2ˆ se cosi’ non fosse
la eq. e le sue soluzioni consentirebbero di distinguere le particelle
Definendo un operatore di Permutazione, cioè di un operatore che scambia le coordinate della particella 1 con quelle della particella 2
( ) ( )122112 ,,ˆ qqqqP Ψ=Ψ
12ˆˆ PH e commutano [ ] 0ˆ,ˆ 12 =PH infatti
( ) 12121221122121121212121212 ˆˆˆˆˆˆˆˆ Ψ=Ψ=Ψ=Ψ=Ψ PHHHHPHP quindi, possono esserci autofunzioni comuni di 12P ed H
q1 e q2 includono coordinate spaziali e di spin
Università di Roma “ La Sapienza” he-17
Dipartimento di Chimica Prof. Guido Gigli
• Vediamo gli autovalori di 12P ( ) ( )21122112 ,,ˆ qqpqqP Ψ=Ψ
( ) ( ) ( )
( ) ( ) ( )
11
,1,ˆ,ˆˆˆ
,,ˆ,ˆˆˆ
12212
2112122112122
12
21212211212211212
212
±==
Ψ⋅=Ψ=Ψ=Ψ
Ψ=Ψ=Ψ=Ψ
pp
qqqqPqqPPP
qqpqqpPqqPPP
quindi
anche ed
• In conclusione per incorporare nella trattazione quantomeccanica la indistinguibilità di due particelle identiche le funzioni d’onda possono essere soltanto quelle che sono autofunzioni simultanee di 12ˆˆ PH e QUINDI
infatti entrambe sono fisicamente possibili perchè la quantità osservabile 2Ψ non varia con lo scambio di particelle
identiche ( 2212ˆ Ψ=ΨP )
qui interviene il Principio di PAULI
POSTULATO Per le particelle con spin intero ( BOSONI ) Ψ=Ψ12P Per le particelle con spin semintero ( FERMIONI ) Ψ−=Ψ12P
• esempi di BOSONI
nucleo di )0(4 =spineH ,nucleo di H2 (spin=1) fotoni
esempi di FERMIONI : Elettroni,Protoni nucleo di eH3
Università di Roma “ La Sapienza” he-18
Dipartimento di Chimica Prof. Guido Gigli
• Una importante conseguenza dell’antisimmetria della Ψ dei fermioni è che:
- se le coordinate di spin e spaziali di due elettroni (1 e 2 ) sono uguali
( ) ( )n4311n4311 qqqqqqqqqq ,,,,,,,,,, KK Ψ−=Ψ il che è vero soltanto se
( ) 0,,,,, 3211 =Ψ nqqqqq K
CIOE’ Due elettroni con il medesimo spin NON possono avere le
stesse coordinate spaziali
Repulsione di PAULI (concetto statistico e non energetico )
• piu’ in generale Bosoni e Fermioni hanno un
diverso comportamento; per esempio, per due particelle identiche e non interagenti che si trovano negli stati quantici a e b:
per Bosoni ( ))2()1()2()1( abbaTot N ψψψψ +=Ψ per Fermioni ( ))2()1()2()1( abbaTot N ψψψψ −=Ψ se ora le due particelle hanno le medesime coordinate
.)()()()1( 2121 eccqqqqqq aaaaa ψψψψψ ====== la densita’ di probabilita’ risultante è ( per Ψ reali )
Bosoni Fermioni
22
222222
2
4
2
ba
bababa
Tot
ψψ
ψψψψψψ
=
++
∝Ψ
0
2 222222
2
=−+
∝Ψ
bababa
Tot
ψψψψψψ
CIOE’
la probabilita’ di essere nel medesimo punto dello spazio aumenta per i Bosoni si annulla per i Fermioni
Università di Roma “ La Sapienza” he-19
Dipartimento di Chimica Prof. Guido Gigli
• Ora dobbiamo integrare i nostri risultati per l’Elio (incompleti, ma non errati) per tenere conto del principio di Pauli
• Anzichè usare la teoria relativistica di Dirac “appiccichiamo” lo SPIN alla teoria di Schroedinger e, quindi, assumiamo che
SO ⋅=⋅Ψ=Ψ (spin)(spaziale) α
ORBITALE
SPIN – ORBITALE • Si può osservare che l’assunzione della Ψ come
prodotto è coerente con la approssimazione non relativistica che non prevede interazione fra lo spin ed il moto della particella nello spazio (anche se già sappiamo che questa interazione, in realtà, esiste; vedi ah 26
• Inoltre è utile notare che ne discende una
conseguenza semplice per le regole di selezione delle transizioni elettroniche
affinchè sia possibile una transizione
=⋅ 2211 ˆ OSOS eµ deve essere diverso da zero quindi
0ˆ 2121 ≠⋅ SSOO eµ il che significa che
021 essere deve ≠SS cioè che
ortogonali essere devono non ed 21 SS
lo Spin non puo’ cambiare
Università di Roma “ La Sapienza” he-20
Dipartimento di Chimica Prof. Guido Gigli
Stato fondamentale dell’Atomo di Elio ( Spin incluso) • Usando le Ψ idrogenoidi, all’ordine zero, la Ψ
(spaziale) è: ( ) ( )2111 ss autofunzione di 12P • Per la Ψ (spin) abbiamo 4 possibilità
tutte autofunzioni di H con il medesimo autovalore
a meno di un termine costante (-1)
=
Università di Roma “ La Sapienza” he-21
Dipartimento di Chimica Prof. Guido Gigli
• In conclusione l’unica accettabile è
( ) ( ) ( ) ( )122143 βαβαφφ −=− complessivamente
( ) ( ) ( ) ( ) ( ) ( )[ ]12212111 βαβα −⋅=Ψ ssN • Notiamo che ➪ la parte spaziale è quella già
considerata ➪ H (nella nostra approssimazione)
non opera sulle funzioni di spin QUINDI
I risultati energetici a cui siamo giunti sono ancora validi
• Si può vedere che questa funzione d’onda è
autofunzione di zSS ˆˆ2 ed con autovalori =0 - Abbiamo ottenuto una sola funzione accettabile - Stato di SINGOLETTO - molteplicità di Spin 112 =+S
Università di Roma “ La Sapienza” he-22
Dipartimento di Chimica Prof. Guido Gigli
• Una interpretazione fisica (da usare con cautela) è che: gli elettroni sono “appaiati” i relativi SPIN giacciono sui “coni” sempre
in direzione opposta
• Infine si può notare che (per H non relativistico
l’operatore Hamiltoniano non opera sulle coordinate di spin)
[ ] [ ] 0ˆ,ˆ0ˆ,ˆ 2 == SHSH z
( zSS ˆˆ2 ed sono costanti del moto ) Donde la convenienza a classificare gli stati con la molteplicità di Spin
Se si introducesse la interazione di SPIN- ORBITA
SOHHH ˆˆˆ +=
[ ] 0ˆ,ˆˆ 2 ≠+ SHH SO
la molteplicità di Spin non sarebbe ben definita compaiono fenomeni nei quali lo Spin cambia
fosforescenza ”intersystem crossing”
( zSS ˆˆ2 ed non sono piu’costanti del moto )
Università di Roma “ La Sapienza” he-23
Dipartimento di Chimica Prof. Guido Gigli
Stato eccitato dell’atomo di Elio Funzioni d’onda
• Con l’approssimazione di totaleΨ prodotto di Ψ idrogenoidi costruiamo delle funzioni d’onda in accordo al principio di Pauli usando ss 21 e
Simmetria Simmetria
( ) ( ) ( ) ( ) ( ) ( )
( ) ( )( ) ( ) ( ) ( )2121
212121122211
αββαββ
αα
+
− ssss
( ) ( ) ( ) ( ) ( ) ( ) ( ) ( )212121122211 αββα −+ ssss • Abbiamo 4 funzioni d’onda: spin totale S 3 stati con la stessa Ψ spaziale TRIPLETTO 1 1 stato SINGOLETTO 0 Dagli autovalori
di 2S • Le Ψ di spin del tripletto corrispondono ad elettroni
per i quali :
➪ 21=sm per entrambi gli elettroni
A
S
S
S
S
A
Parte Spaziale
Parte di Spin
Università di Roma “ La Sapienza” he-24
Dipartimento di Chimica Prof. Guido Gigli
➪ 21−=sm per entrambi gli elettroni
➪ sm � “miscela” simmetrica di 2
1 e 21−
• Le Ψ di spin del singoletto corrispondono ad elettroni
per i quali : ➪ sm � “miscela” antisimmetrica di 2
1 ed 21−
Energie • Per vedere quale sia l’energia corrispondente a questi
nuovi “stati” ( funzioni d’onda ) possiamo calcolare il valore di aspettazione di
ssJ 21 ⇒ Integrale di Coulomb ⇒ repulsione fra le densita’ di carica associate agli 1s e 2s
infatti
[ ] [ ] 2112
2221
1)2(2)1(1 ττ ddr
ssJ ss ∫∫=
⇒ è un termine sempre positivo che per lo He vale .4195 Hartrees
sono identici a coppie in quanto è indifferente etichettare gli elettroni come 1 e 2 oppure 2 ed 1 ( gli indici sono fittizi perchè la integrazione è sulle coordinate sia di uno che dell’altro elettrone )
Università di Roma “ La Sapienza” he-26
Dipartimento di Chimica Prof. Guido Gigli
ssK 21 ⇒Integrale di scambio
⇒ ha origine dal requisito di antisimmetria della totΨ
⇒ rappresenta la repulsione fra la
sovrapposizione ss 21 e se stessa
[ ] [ ] [ ][ ] 2112
21 )2(2)2(11)1(2)1(1 ττ ddssr
ssK ss ∫∫=
⇒ normalmente (ma non sempre) positivo (
per lo Hartrees044.=eH ) ⇒ fisicamente si può osservare che la
funzione d’onda si annulla se gli elettroni hanno le stesse coordinate
⇒ poiché compare con un segno negativo
(ed è di norma positivo) la energia è minore per una Ψ antisimmetrica di quanto sarebbe stata per una Ψ di elettroni distinguibili
⇒ in quanto è antisimmetrica la Ψ si
annulla se gli elettroni hanno le medesime coordinate: Il “buco” (buca di Fermi) che ne risulta diminuisce la repulsione ed abbassa l’energia totale
⇒ si può anche notare che poiché la Ψ è continua e deve divenire nulla per q(1)=q(2) essa deve incominciare a diminuire prima che le coordinate coincidano
Università di Roma “ La Sapienza” he-27
Dipartimento di Chimica Prof. Guido Gigli
• Complessivamente
HartreesHartrees
EEEEE hhhh
1755.21245.2
044.04195.0212
−↔−=
=−+−−=
sperimentale Stato di Singoletto
HartreesHartrees
KJEEE ssssss
1465.20365.2212121
−↔−=
=+++=
sperimentale ETripletto < ESingoletto DEcalc ( 0.088)= buon successo = 3.03 DEsperim (0.029) MA
Università di Roma “ La Sapienza” he-28
Dipartimento di Chimica Prof. Guido Gigli
Determinanti di Slater • Il modo usato per ricavare le Ψ correttamente
antisimmetrizate è troppo laborioso per atomi con più elettroni
• Slater ha introdotto un metodo che sfrutta le
proprietà dei determinanti di cambiare di segno scambiando righe o colonne
• Consideriamo, per esempio, uno degli stati di
tripletto dell’Elio ( ) ( )[ ]12m1m ss =+
( ) ( ) ( ) ( )[ ] ( ) ( )[ ]
( ) ( ) ( ) ( )( ) ( ) ( ) ( )
( ) ( )( ) ( )
( ) ( )( ) ( )2SO2SO
1SO1SON
2SO2SO1SO1SO
N
22s222s111s211s1
N
212s11s22s21s1N
21
21
21
21
⋅=
=⋅=
=⋅=Ψ
⋅−⋅=Ψ
αααα
αα
• Un’altra notazione usata frequentemente è quella
di scrivere soltanto gli elementi della diagonale della matrice
• Notiamo che : Scambiando righe o colonne la Ψ cambia di segno
La Ψ è correttamente antisimmetrica rispetto allo scambio di elettroni
➭Se due righe fossero uguali (OS1 =OS2 ) la Ψ si annullerebbe
non esiste la Ψ corrispondente a due elettroni assegnati al medesimo Spin-Orbitale
in accordo con il Principio di Esclusione di Pauli
Due elettroni ( Due Fermioni ) non possono essere simultaneamente nel
medesimo stato quantico ( avere lo stesso insieme di numeri quantici )
Università di Roma “ La Sapienza” he-30
Dipartimento di Chimica Prof. Guido Gigli
• Le cose non sono sempre così semplici; si deve ricorrere a:
combinazioni di Det. di Slater (per
atomi con shell non complete) Teoria dei gruppi
INOLTRE
• Stiamo pur sempre usando le approssimazioni:
Particelle non interagenti Funzioni d’onda monoelettriche Spin – Orbitali
IN REALTA’
• La eq. di Schroedinger per molti elettroni non è
separabile QUINDI
La Ψ totale non può essere scritta come un prodotto ( anche se antisimmetrizzato ) di
funzioni monoelettroniche IN SINTESI • Con i Determinanti di Slater:
NON violiamo il principio di Pauli
NON abbiamo risolto del tutto il problema: in
pratica lo scambio di elettroni non è un fenomeno fisico ma soltanto la correzione di un errore di origine e cioè della errata descrizione che no tiene conto della indistinguibilita’ degli elettroni.
La correlazione fra il moto degli elettroni non viene pienamente considerata
Università di Roma “ La Sapienza” ap-1
Dipartimento di Chimica Prof. Guido Gigli
ATOMI POLIELETTRONICI
• Vediamone dapprima una descrizione qualitativa rimandandone i dettagli quantitativi.
• Non è possibile risolvere la eq. di Schroedinger per sistemi ( atomi o molecole ) con due o più elettroni
• Ci si aspetta, comunque, che si possa ancora usare la approssimazione di funzioni monoelettroniche, cioè di orbitali che, pur essendo diversi da quelli degli atomi idrogenoidi, siano in numero uguale ed abbiano dipendenze angolari simili.
• Si possono,allora, usare degli orbitali idrogenoidi “distorti” per descrivere la struttura elettronica di un atomo con più elettroni.
• Il campo elettrico prodotto dagli altri elettroni, però, rimuove la degenerazione degli orbitali con lo stesso numero quantico principale
• La sequenza dei livelli energetici diviene allora la seguente ( si può det. sperimentalmente )
.....43433221 pdspspss <<<<<<<
Università di Roma “ La Sapienza” ap-2
Dipartimento di Chimica Prof. Guido Gigli
• La situazione può essere descritta schematicamente come nel diagramma che segue
• L’ordine degli orbitali dipende,inoltre, dal numero atomico
Università di Roma “ La Sapienza” ap-3
Dipartimento di Chimica Prof. Guido Gigli
Penetrazione e “Shielding”
• Il quesito è: perchè gli orbitali “p” hanno energia maggiore degli “s” ?
• Negli atomi polielettronici un elettrone risente di
• attrazione del nucleo
• repulsione degli altri elettroni
L’effetto complessivo è che l’elettrone “vede”una carica nucleare effettiva Zeff = Z - S
Shielding Costante di schermo
• La differenza nella parte radiale delle funzioni d’onda significa una diversa
Penetrazione
• L’elettrone “s” può trovarsi sul nucleo (il “p” no )
• L’elettrone ,per esempio,2s “penetra” attraverso gli elettroni 1s che “schermano” gli elettroni esterni e risente in misura maggiore del 2p della carica nucleare
• L’effetto combinato della “penetrazione” e dello “schermo” separa le energie degli elettroni 2s e 2p
• Si tratta ora di vedere con quali regole si possono “assegnare” gli elettroni agli orbitali
Principio di Esclusione di Pauli
Università di Roma “ La Sapienza” ap-4
Dipartimento di Chimica Prof. Guido Gigli
• Ogni orbitale è in grado di “ospitare” soltanto due elettroni (uno con 2121 −== ss mm conunoed )
• Si tratta,allora, di “riempire” la sequenza degli orbitali seguendo il principio dello AUFBAU
Si dispongono gli elettroni negli orbitali ( non più di due elettroni per orbitale) cominciando da quelli ad energia minore in ordine di energia crescente
• Si ottengono le cosiddette configurazioni
Na ∧ Na I 1s2 2s2 2p6 3s
Cl ∧ Cl I 1s2 2s2 2p6 3s2 3p5
Al+ ∧ Al II 1s2 2s2 2p6 3s2
• Si deve notare che esistono configurazioni meno stabili
Na I Ne 3p (a ~ 16960 cm-1 dal fondamentale)
Na I Ne 4s (a ~ 25700 cm-1 dal fondamentale)
• Per ogni configurazione si possono avere vari stati di moto con energia diversa
Termini e livelli spettroscopici
Università di Roma “ La Sapienza” ap-5
Dipartimento di Chimica Prof. Guido Gigli
• E’ il principio di Esclusione che porta all’esistenza stessa della tavola periodica degli elementi ed alle conseguenti PERIODICITA’
• Per esempio per le Energie di Ionizzazione
èlungo un periodo la carica nucleare aumenta e,quindi, aumenta l’energia di interazione nucleo-elettroneLi - Be , B -C - N , O - F - Ne
è Gli orbitali p sono meno legati degli s ecc.Be - B
è quando l’”Aufbau” porta ad accoppiare gli elettroni la repulsione fra questi è maggiore di quanto ci si attende da una semplice estrapolazione
N -O
è Un cambio di numero quantico principale significa una maggiore distanza ed un efficiente “schermo” da parte del guscio completo del periodo precedenteNe - Na
Università di Roma “ La Sapienza” ap-6
Dipartimento di Chimica Prof. Guido Gigli
• Vediamo,ora, di affrontare il problema degli atomi polielettronici in modo più rigoroso.
• L’operatore Hamiltoniano, supponendo il nucleo fermo, è il seguente:
∑ ∑ ∑∑>
+−∇
−=i i ij ijii
irr
ZH 12
2
• La vera autofunzione dovrà quindi essere una Ψche tiene conto complessivamente delle coordinate di tutti gli elettroni
( )nnn z,y,x.,..y,x,z,y,x 22111Ψ=Ψ
Il moto di ogni elettrone dipende dal moto degli altri in quanto ognuno risente della posizione relativa
degli altri attraverso il termine repulsivo
ijr1
- Proprio questo termine impedisce di fattorizzare il problema , separare le variabili e risolvere la eq. di Schroedinger.
- Non si puo’ nemmeno trattare il tutto perturbativamente perchè il contributo di questi termini è dello stesso ordine di grandezza degli altri
• Si deve ricorrere a metodi approssimati
METODO HF-SCF
Università di Roma “ La Sapienza” ap-7
Dipartimento di Chimica Prof. Guido Gigli
Metodo HF-SCF ( Self Consistent Field )Campo Auto Coerente
Auto Consistente
• Dobbiamo usare la approssimazione monoelettronica e, quindi, continuare ad usare gli “spin-orbitali”
MApossiamo scrivere la funzione d’onda poliettronica in accordo al principio di Pauli come prodotto antisimmetrizzato di funzioni monoelettroniche (determinante di Slater)
dove Oi è un orbitale (funzione delle sole coordinate spaziali)
Oiα è uno spin orbitale (funzione delle coordinate spaziali e di quella di spin)
• In questo modo assumiamo di poter descrivere la struttura elettronica con un unico determinante con orbitali tutti doppiamente occupati
stiamo usando la forma piu’ semplice del metodo (valida per shell “chiuse”, stati di singoletto)
Università di Roma “ La Sapienza” ap-8
Dipartimento di Chimica Prof. Guido Gigli
• L’idea è quella di cercare la miglior forma analitica delle funzioni spaziali monoelettroniche Oi con il metodo variazionale.
si cerca, quindi, il minimo del valore di aspettazione dell’energia della funzione scritta come determinante
DetHDetE =detto in altri termini si usa come funzione di prova il determinante di Slater degli orbitali monoelettronici
• Hartree e Fock hanno dimostrato che si ottiene, per ogni orbitale, una equazione, cosiddetta di Hartree Fock di forma identica alla eq. di Schroedinger:
)elettroninumeron;2nN,1,2,i( OOF iiii ====ε= K
Il singolo ORBITALE SPAZIALE
[ ]KJrZ
VrZ
n
jjj
i
i
HFi
i
i
−+−∇
−
=
+−∇
−
∑=
2
1
2
2
22
2
Operatore di Fock(Hamiltoniano Effettivo)
Autovalore dell’energia dell’ORBITALE
Università di Roma “ La Sapienza” ap-9
Dipartimento di Chimica Prof. Guido Gigli
• In questo operatore sono identificabili:
• due termini esatti relativi a
- energia cinetica
- potenziale nucleo - elettrone
• termini approssimati relativi alle interazioni elettrone-elettrone
operatore di Coulomb jJ
§ ( ) ( ) ( ) ( ) ( )12121112
ijjij OOrOOJ =
da notare che - gli indici i e j identificano gli orbitali (per
esempio 1s, 2p ecc.)- 1 e 2 indicano la generica coppia di
elettroni che si sta considerando- l’integrazione è fatta sulle coordinate
spaziali dell’elettrone 2 nell’orbitale j§ il singolo termine della sommatoria
rappresenta il potenziale repulsivo medio e locale, nella posizione assunta dall’elettrone 1 nell’orbitale i , generato dall’elettrone 2 nell’orbitale j.
§ È la parte di operatore da cui si originano, nella energia, questi termini repulsivi
( ) ( ) ( ) ( )
12
22
21
12
21
21121
r)(O)(O
dd
OOrOOOJOJ
ji
jijiijiij
ττ=
===
∫∫
in tutto simili all’integrale di Coulomb gia’ visto per l’atomo di Elio
Costituiscono il cosiddetto
Hamiltoniano di “core” nel senso che, per ciascun elettrone, non tengono conto
degli altri
Università di Roma “ La Sapienza” ap-10
Dipartimento di Chimica Prof. Guido Gigli
§ Dalla forma dell’operatore si vede che dipende da tutti gli altri elettroni che occupano gli orbitali j mediati su tutto il volume
§ è un operatore cosiddetto locale perché il suo effetto sull’orbitale Oi dipende dal valore dell’orbitale a ciascuna specifica coordinata assunta dall’elettrone che lo occupa
operatore di Scambio jK
( ) ( ) ( ) ( ) ( ) ( ) ( ) ( )1221212111212
ijij
jjijij OOrP
OOOrOOK ==
non ha analogo classico perché ha origine dalla antisimmetrizzazione della funzione d’onda complessiva e dalla presenza nell’Hamiltoniano dei termini di interazione
ijr1§ ne derivano, nell’energia, termini simili
all’integrale di scambio già visto per l’atomo di Elio (potenziale di scambio)
( ) ( ) ( ) ( )
( ) ( ) ( ) ( )12
21
12
2211
21121
rOOOO
dd
OOrOOOKOK
i*jj
*i
ijjiijiij
ττ=
===
∫∫
§ )i(K j è un operatore non locale perché non rappresenta un semplice potenziale definito univocamente nella posizione dell’elettrone 1. Il suo effetto quando è applicato all’orbitale Oi non dipende dal valore di Oi ad uno specifico valore della coordinata assunta dall’elettrone ma dal valore di Oi in tutto lo spazio: in effetti, a causa dello
Università di Roma “ La Sapienza” ap-11
Dipartimento di Chimica Prof. Guido Gigli
scambio, l’orbitale Oi è ora all’interno dell’integrale
• nell’operatore di Fock
[ ]∑=
−+−∇
−=+−∇
−=2
1
22222
n
jjj
i
iHFi
i
ii KJr
ZVrZF
si puo’ notare che:§ gli operatori di Coulomb e di Scambio vanno
a costituire un potenziale “centrale” HFiV di
cui risente l’operatore i-esimo§ l’operatore di Coulomb compare con un
coefficiente 2 perchè va considerata la repulsione di entrambi gli elettroni dell’orbitale j
§ l’operatore di scambio, viceversa, ha effetto soltanto per uno degli elettroni nell’orbitale j(quello con il medesimo spin dell’elettrone nell’orbitale i che si sta considerando); l’altro possibile termine di scambio si annulla per l’ortogonalità delle funzioni di spin
• Sia il termine di Coulomb che quello di Scambio dipendono dalla conoscenza degli altri orbitali che, all’inizio del calcolo, sono sconosciuti. Addirittura l’operatore di Fock ha la peculiare caratteristica di dipendere dalla sua stessa autofunzione
( ) ( ) ( ) ( ) ( )112211 212
ji*jij Od
rOOOK ⋅
τ= ∫
non note “a priori”
QUINDIper esempio, per l’elettrone 1 abbiamo
1111 OOF ε=
Università di Roma “ La Sapienza” ap-12
Dipartimento di Chimica Prof. Guido Gigli
•questa equazione può essere impostata e risolta se si conoscono le jO di tutti gli altri elettroni
• Il problema è circolare e viene risolto in modo ITERATIVO
1. ) Si risolve (numericamente) 1111 OOF ε=ipotizzando le altre funzioni jO
2. ) con la nuova 1O si passa ad 2222 OOF ε=
- Si ripete il passo 2) sino all’elettrone n- Si continua sino a che la totΨ non differisce
da quella precedente entro limiti prefissati
• In conclusione si ottengono funzioni monoelettroniche autocoerenti che sono corrette per l’effetto dovuto all’interazione media tra gli elettroni
• Da notare che :
1. ) Il potenziale usato è di tipo a simmetria centrale e la parte angolare delle Ψrisultanti è ancora quella di un atomo idrogenoide ; in effetti per la parte angolare delle Ψ vengono usate le armoniche sferiche
( ) ( )ϕϑ= ,YrfO lmi
ed è soltanto la parte radiale che viene variata nella procedura di ottimizzazione SCF
Si può mantenere la classificazione in orbitali s, p, d ecc.
Università di Roma “ La Sapienza” ap-13
Dipartimento di Chimica Prof. Guido Gigli
2. ) Anche se la parte radiale non è più quella degli atomi idrogenoidi (il potenziale non è piu’ soltanto il pot. di interazione nucleo-elettrone ) il numero di “nodi” radiali viene conservato
Si può continuare ad individuare gli orbitali come 1 s, 2 s ecc.
• In sintesi le equazioni HF
iiii OOF ε=costituiscono un insieme di pseudo-equazioni agli autovalori in quanto l’operatore di Fock dipende dalle soluzioni stesse delle equazioni
• Per quanto riguarda l’energia totale essa non è la somma delle energie dei singoli orbitali
∑=
ε≠2
12
n
iitotE
perché in ciascuna di queste è già inclusa la interazione dell’elettrone i con tutti gli altri (interazione che è stata calcolata con gli operatori
jJ e jK )
Università di Roma “ La Sapienza” ap-14
Dipartimento di Chimica Prof. Guido Gigli
• La somma delle energie dei singoli orbitali va quindi corretta per evitare di contare due volte le interazioni interelettroniche
( )∑ ∑∑ −−ε==
2 22
122
n
i
n
jijij
n
iitot KJE
• Infine ciascuna energia iε , con l’approssimazione detta di Koopmans, viene considerata pari all’energia necessaria per rimuovere l’elettrone dall’orbitale i nell’ipotesi che le funzioni che descrivono il moto degli altri elettroni non subiscano modifiche
Si trascura, quindi, il riarrangiarsi del moto degli elettroni residui (si dice che se ne trascura il “rilassamento”)
• Tornando agli orbitali la cui forma analitica viene ottimizzata variazionalmente, essendo numerica la soluzione delle equazioni di Fock, le funzioni d’onda risultanti, in origine, non venivano rappresentate analiticamente ma mediante tabelle numeriche
• Roothaan (1951) introdusse la rappresentazione degli orbitali HF come combinazioni lineari di un insieme completo di funzioni note
Funzioni di BaseSet di Base
( in generale l’orbitale è espresso come una espansione
∑ ϕ=k
kki cO )
Università di Roma “ La Sapienza” ap-15
Dipartimento di Chimica Prof. Guido Gigli
• Il vantaggio del metodo è che i coefficienti ( kc ) si trovano iterativamente usando l’algebra delle matrici ( adatta ai calcolatori ) e che la forma anlitica delle funzioni di base non viene modificata
• La scelta del “set di base” è libera
• Il metodo è AB INITIO nel senso che si fa uso soltanto dei principi primi e non di parametri “adattabili” ai casi concreti
• Un insieme di funzioni usato come “ set di base”, e che ha un chiaro significato fisico, è quello degli
Orbitali Tipo Slater S T O
( ) ( )φθξ
,,01
mlar
n YerN−
−
da notare la somiglianza con le Ψ idrogenoidi. Il polinomio di grado n-1 viene sostituito con il solo termine “più pesante“ ( r n-1 )
• Attualmente, per rendere piu’ facile il calcolo degli integrali, si preferiscono, prevalentemente, “set” di base che fanno uso di varie funzioni gaussiane la cui combinazione cerca di simulare gli orbitali STO
1s STO
3 gaussiane
1 gaussiana
Università di Roma “ La Sapienza” ap-16
Dipartimento di Chimica Prof. Guido Gigli
• I risultati dei calcoli HF riproducono, e ne forniscono una giustificazione per l’uso, la nota distribuzione a “ gusci “ della densità elettronica radiale
• Anche dal punto di vista quantitativo i risultati HF sono in accordo con le esperienze
- per la funzione d’onda ( Densità elettronica radiale )
- per le energie
He Stato fondamentaleHF -77.9 eV
“vero” (nonrelativistico) -79.0 eV
1 s HF -25.0 eVPotenziale di Ionizzazione 24.6 eV
Li Stato fondamentaleHF -202.3 eV“vero” -203.5 eV
Università di Roma “ La Sapienza” ap-17
Dipartimento di Chimica Prof. Guido Gigli
• Anche le singole energie di ionizzazione e la periodicità di queste sono ben riprodotte
Energia di ionizzazione/MJ mol-1
elettrone orbitali app. di calcolirimosso risultanti Koopmans HF sperimentale
• In linea di principio si dovrebbe usare un numero infinito di funzioni di base
in pratica se ne usa un numero finito
• Per calcoli o rappresentazioni più grossolane è scomodo usare la combinazione di molti STO
Slater propose di usare una singola funzione ( STO ) dove
===
−=
quanticonumeroilconvariacheparametro
schermodicostanteatomiconumeroZ
** n
Sn
SZξ
1* −nr
Questi parametri sono ricavabili con regole fornite originariamente dallo stesso Slater ed in seguito modificate
Università di Roma “ La Sapienza” ap-19
Dipartimento di Chimica Prof. Guido Gigli
Correlazione Elettronica
• Le Ψ HF – SCF tengono conto della interazione fra gli elettroni in modo mediato
Dovremmo considerare la interazione istantanea
infatti il moto degli elettroni è correlato
• In una qualche misura anche le Ψ HF – SCF tengono conto della correlazione in quanto soddisfano il principio di Pauli
Le Ψ vanno a zero per elettroni con il medesimo spin che si trovano nello stesso punto dello spazio(nelle Ψ HF-SCF si può parlare di Buca di Fermi )
• La Energia di Correlazione viene definita come
HFlnon esattaCorr EEE −= ..Re
2
Re10−<
.lnon esatta
CorrE
E HFE pressochè corretta
MA
totE è grande cE =-1000 eV
≅CorrE 5 eVCOME I
LEGAMI CHIMICI
Università di Roma “ La Sapienza” ap-20
Dipartimento di Chimica Prof. Guido Gigli
• Per tener conto della correlazione istantanea• si introduce esplicitamente la distanza interelettronica nella funzione d’onda
• si usa il metodo CI (ConfigurationsInteraction )
∑=Ψi
ii DC
iD sono i determinanti di Slater per- stato fondamentale- stati eccitati di§ 1 elettrone§ 2 elettroni§ tutti “ (FCI)
Full CI
• I coefficienti iC possono essere ottimizzati con il metodo variazionale
• Numericamente il problema diventa rapidamente non facile da trattare ( Determinanti enormi )
• Se si usasse
• un set base completo• tutti i possibili iD
Il metodo sarebbe ESATTO HF – FCI
Università di Roma “ La Sapienza” ap-21
Dipartimento di Chimica Prof. Guido Gigli
Costanti del Moto
• Ci siamo occupati di trovare, per quanto possibile, autofunzioni ed autovalori di H
• Per l’atomo idrogenoide avevamo visto che le classificazioni degli elettroni e delle configurazioni era fatta sulla base di numeri quantici che governavano altre caratteristiche ( oltre la Energia Totale ) del moto degli elettroni ( Momento angolare totale, componente lungo un asse del Momento Angolare )
Quello che abbiamo fatto,in linea di principio, è stato interessarci di altre
costanti del moto
cioè di altri autovalori di operatori che commutano con H
INFATTI
In questa ipotesi la Ψ per cui Ψ=Ψ EH è anche quella per cui Ψ=Ψ aA a ed i numeri quantici
in a non dipendonodal tempo
Università di Roma “ La Sapienza” ap-22
Dipartimento di Chimica Prof. Guido Gigli
• Si dice che questi numeri quantici sono:
Buoni Numeri Quantici
per la classificazione degli elettronisiano interessati ad individuarli
• Quando un operatore “ quasi commuta “ con H si possono avere numeri quantici “quasi” buoni
• tipicamente 10ˆˆˆ HHH λ+=
[ ] [ ] 0ˆ,ˆ0ˆ,ˆ10 ≠= HAHA ma
• per λ piccolo i numeri quantici associati ad Avariano poco con il tempo e sono quasi buoni
• Si può vedere che:
• Senza l’interazione SPIN-ORBITA
H commuta con operatori composti relativi al momento angolare di Spin di tutti gli elettroni
Krrr
Krrr
++=++= 21 SSS 21 LLL
abbiamo dei buoni numeri quantici L ed S
• Con l’interazione SPIN-ORBITA
H commuta soltanto con
+=
+=++=
222
11121
SLJ
SLJJJJ rrr
rrr
Krrr
• Si usano QUINDI due schemi:
♦ RUSSEL SAUNDERS
♦ ACCOPPIAMENTO JJ Modello Vettoriale dell’ Atomo
Università di Roma “ La Sapienza” ap-23
Dipartimento di Chimica Prof. Guido Gigli
Modello Vettoriale – Accoppiamento RUSSEL-SAUNDERS• Il primo metodo ( che vedremo in dettaglio ) è
adatto per gli atomi leggeri mentre il secondo lo è per quelli più pesanti
• I vettori JSLrrr
e,, rappresentano delle entità trattabili con la quantomeccanica per le quali
( )( )( )
quanticinumerisonoJSL
JJJ
SSS
LLL
,,
1
1
1
+=
+=
+=
hr
hr
hr
• Di nuovo si possono usare vari metodi per ricavare le somme vettoriali ed i numeri quanticiUsiamo il metodo della somma delle componenti
• Consideriamo,per esempio, due elettroni ( l1=2, l2=1)
l1=2 +2 +1 0 -1 -2 = ml1=l1z/h
l2=1 +1 0 -1 = ml2=l2z/h
combinando ciascuno degli ze lm o si ha∑∑ == i zizi ilL lLmM
infatti si può costruire una tabella
L ML
3 +3 +2 +1 0 -1 -2 -32 +2 +1 0 -1 -21 +1 0 -1
da cui ( ) 212121 ,,1,,2,1,
llllllLLLLLLM zL
−−++=
=−−−+=
K
hK
Serie di Clebsch-Gordan (soltanto per due elettroni)
Università di Roma “ La Sapienza” ap-24
Dipartimento di Chimica Prof. Guido Gigli
• Analogamente per S ed Sz si ha
∑=−−==
iisS
zS
mMSSSSM Kh 1,/
• Per J e Jz = Lz + Sz
( ) SLSLSLJJJJJM zJ
−−++=
−−==
,1,,1,/ h
• Ora siamo in grado di scrivere i simboli spettroscopici per atomi polielettronici secondo la solita convenzione
JS L12 + L = 0 1 2 3
S P D F
• Consideriamo, per esempio, l’atomo di litioconfigurazione ss 21 2
• Possibili valori di ML ? 0000321 =++=++= lllL mmmM
• valori possibili di L ? (ML = L, L-1 -L)00 =→= LM L
• valori di MS ?
21,21212121321
−+=±−+=++=
S
sssS
MmmmM
• valori di S ? (MS = S, S-1, -S )2121,21 =→−= SM S
Serie di Clebsch-Gordan
Università di Roma “ La Sapienza” ap-25
Dipartimento di Chimica Prof. Guido Gigli
quindi per ss 21 2 si ha
212
2
21
211,
210
S
SL
SLSLSLSLJS
SL
=−
=+−−++=
==
......
• Per la configurazione eccitata ps 21 2
110010000
1100
−=−+==++
=++=L
M L
21
2121
21,
21,
21
=
−
+=±−+= SM S
232
212
2
21,23
PP
JP
=
• Si ha ancora la “gerarchia”
Configurazione psss 2121 22
Termine 2S 2PLivello 23
221
221
2 PPS
Stato ( per esempio ) JMS ed212→ specificato
Università di Roma “ La Sapienza” ap-26
Dipartimento di Chimica Prof. Guido Gigli
• Consideriamo l’atomo di Carbonioconfigurazione 222 221 pss
E’ utile costruire una tabella dei valori possibili di SL MM ed ( è sufficiente considerare gli elettroni
in shells non completamente riempite )• Gli elettroni p hanno 1,0,1 −+=lm
ed 21,21 −+=sm
Il campo dei valori possibili di ML è +2 ÷ -2 e di MS è 1 ,0 ,-1
• Riempiamo le caselle della Tabella con tutte le combinazioni possibili (permesse dal principio di esclusione ) ( )21,21 −→−=+→+= ss mm e deriviamo i simboli dei termini
−+
−−+−−+++
−+
−−+−−+++
−−+−−+++
−+++
−−−
−−−−−
−−−−
−
1,12
0,10,10,10,11
0,0
1,11,11,11,10
0,10,10,10,11
1,11,12
101
L
S
MM
3PL = 1 S=1MS = 1,0,-1ML = 1,0,-1
9 microstati
1DL = 2 S = 0
5 microstati
1SL = 0 S = 0
1 microstato
Università di Roma “ La Sapienza” ap-27
Dipartimento di Chimica Prof. Guido Gigli
• Quindi dalla configurazione 2p2 si originano i tre termini
• vediamo meglio, con un esempio, perché differenti termini e differenti livelli hanno energie diverse:
§ gli stati di moto −+
−1,1 (del 3P) ed −+1,1
(del 1D) hanno energia diversa perché c’è una diversa repulsione interelettronica in quanto i due elettroni sono, in un caso, nello stesso orbitale e, nell’altro, in orbitali diversi
§ gli stati di moto −+
−1,1 (del 3P) ed ++
−1,1 (del 3P) hanno energia diversa a causa della differente entita’ della interazione spin-orbita
QUINDI
§ i TERMINI differiscono per la repulsione interelettronica
§ i LIVELLI differiscono per un diverso accoppiamento spin-orbita
Università di Roma “ La Sapienza” ap-28
Dipartimento di Chimica Prof. Guido Gigli
configurazione termini livelli stati
1S 1S0
1D 1D2
3P2
3P1
3P1 p2
• Per sapere come sono disposti in energia questi livelli si usano le regole di HUND
1 ) A parità di configurazione il livello più stabile è quello a maggiore molteplicità di spin ( S più grande)
2 ) Per livelli con pari S è più stabile quello con L maggiore
3 ) In relazione al J : quando una shell è riempita meno della metà i livelli più stabili sono quelli con J più basso e viceversa
• Nel caso del Carbonio le regole funzionano, infatti:
01 S 21648.4 cm-1
21 D 10193.7 cm-1
23 P 43.5 cm-1
13 P 16.4 cm-1
03 P 0 cm-1
Università di Roma “ La Sapienza” ap-29
Dipartimento di Chimica Prof. Guido Gigli
Università di Roma “ La Sapienza” mb-1
Dipartimento di Chimica Prof. Guido Gigli
MOLECOLE • Vedremo come la meccanica quantistica spiega la
formazione di un legame stabile fra gli atomi • Lo ione molecolare +
2H sarà il nostro “modello” per molecole più complicate
così come l’ Aufbau ci ha consentito di comprendere gli atomi polielettronici collocando elettroni ( in accordo al principio di esclusione di Pauli ) negli orbitali trovati per l’atomo più semplice (Idrogeno) ,la più semplice molecola possibile ( +
2H ) ci fornirà le funzioni d’onda monoelettroniche adatte a costruire gli orbitali molecolari Eq. di S.per H Eq. di S. per +
2H Orbitali Idrogenoidi Orbitali di +
2H Determinanti di Funzioni d’onda di Slater molecole
Università di Roma “ La Sapienza” mb-2
Dipartimento di Chimica Prof. Guido Gigli
Approssimazione di Born-Oppenheimer La vediamo per lo +
2H ma è generalizzabile • Hamiltoniano di +
2H e ra rb a rab b
=H b
b
a
amm 22
22 ∇−∇− 2
2e∇−
ba rr11 −−
abr1+
nT eT enV nnV
( ) =Ψ en qqH ,ˆ ( )en qqE ,Ψ • Il problema è a 3 corpi → la eq. di Schroedinger
non è fattorizzabile → possiamo comunque separare il moto del baricentro dai moti interni (nuclei ed elettroni rispetto al baricentro)
• se possiamo trascurare nT l’hamiltoniano si riduce a: eT + enV + nnV
• In effetti il problema trae vantaggio dalla
osservazione che
massa nuclei >> massa elettroni
• Gli operatori di energia cinetica dei nuclei sono assai piu’ piccoli di quelli degli elettroni
Università di Roma “ La Sapienza” mb-3
Dipartimento di Chimica Prof. Guido Gigli
• Inoltre, se anche la dipendenza della funzione d’onda dalle coordinate dei nuclei e degli elettroni e’ sensibilmente diversa, ne consegue che:
• I nuclei sono pressochè fermi nel tempo
caratteristico del moto degli elettroni
Possiamo risolvere il problema elettronico, cioè del moto dei soli elettroni, con i nuclei in posizioni
fissate ( ) Ψ⋅=Ψ+ elene EVT
• la nostra Ψ dipenderà dalla posizione dei soli elettroni nell’approssimazione che i nuclei siano fermi
• peraltro vi sarà una Ψ per ogni singola distanza fra i nuclei
Quindi avremo una funzione d’onda siffatta:
( )Rr,Ψ r coord. di tutti gli elettroni R coord. di tutti i nuclei
( )Rr;Ψ - dipende dalla posizione degli
elettroni in modo esplicito (analitico) -dipende dalla distanza fra i nuclei in modo parametrico nel senso che
una volta fissata non vi dipende piu’
• Risolto il problema elettronico (per tutte le distanze fra i nuclei) si potrà affrontare quello dei moti dei nuclei
( ) ( ) ( )REREVT elnnn χχ =++ ˆˆ
Università di Roma “ La Sapienza” mb-4
Dipartimento di Chimica Prof. Guido Gigli
• in conclusione abbiamo due eq. di Schroedinger
( ) Ψ=Ψ+ elene EVT
( ) χχ EEVT elnnn =++ˆ
( ) χχ EVT nn =+ˆ
e’ per tutto cio’ che si puo’ usare una curva di potenziale come quella di MORSE
Vr
R
QUINDI • Il nostro vero problema è risolvere la eq. di
Schroedinger elettronica ( ) Ψ=Ψ+ elene EVT
INFATTI
Il termine ab
nn rV 1+= si può facilmente aggiungere ad
elE per calcolare elnn EV +
• notiamo che, cosi’ come negli atomi, avremo vari stati elettronici (stati di moto degli elettroni)
Eq. di S. ELETTRONICA
le coordinate nucleari sono soltanto parametri
Eq. di S. NUCLEARE
Superficie ( Curva ) di Energia Potenziale ( anche se oltre al vero Potenziale Vn il termine Eel include la energia cinetica degli elettroni )
Università di Roma “ La Sapienza” mb-5
Dipartimento di Chimica Prof. Guido Gigli
Molecola Ione +2H
• Con l’approssimazione di Born-0ppennheimer la
eq. elettronica è:
=Ψ
−−∇−ba rr11
2
2 ΨelE
Esattamente risolubile in Coordinate ellittiche ( )φηξ ,,
angolo di Rotazione intorno all’asse di legame
Due distanze
ab
barrr +=ξ
ab
barrr −=η
• In queste coordinate il problema è fattorizzabile e le
autofunzioni sono un prodotto di funzioni:
( ) ( )( )
φ
πηξ im
el eML 2121=Ψ
• Per la Energia: nel punto di minimo
1033.1Eel −= 6026.0ETOT −=
TOT
e
ED
%171026.0
==
Università di Roma “ La Sapienza” mb-6
Dipartimento di Chimica Prof. Guido Gigli
• Piu’ complessivamente:
Università di Roma “ La Sapienza” mb-7
Dipartimento di Chimica Prof. Guido Gigli
• Notiamo che:
Le varie curve sono indicate con simboli diversi
gug πσσ ,, INFATTI
mentre per l’atomo di Idrogeno la simmetria sferica del potenziale faceva sì che H commutasse
con 2L e con zL ora soltanto zL commuta con H
Il momento orbitale elettronico totale
non è una costante del moto lo è soltanto la sua componente
• Di norma si usa un simbolo per indicare il valore di
λ=m 43210λ
σ π δ φ γ Simmetria cilindrica lungo il legame • Si indica anche la parità della Ψ
Ψ pari gerade g gσ
Ψ dispari ungerade u uσ
Università di Roma “ La Sapienza” mb-8
Dipartimento di Chimica Prof. Guido Gigli
Trattazione approssimata di +
2H
Una osservazione sul metodo • Le soluzioni esatte per lo +
2H sono non facilmente “maneggiabili” e di difficile interpretazione fisica
• Possiamo seguire una via simile a quella degli atomi polielettronici
• Per questi ultimi abbiamo usato il metodo SCF per costruire Ψ approssimate come Det. di Slater di spin orbitali monoelettronici la cui parte spaziale sono gli orbitali atomici
( ) mlYrR ,
una buona app. iniziale sono funzioni radiali con cariche nucleari efficaci
• Per le molecole possiamo usare:
Ψ approssimate come Det. di Slater di spin orbitali monoelettronici ↑ la parte spaziale ⇔ Orbitali Molecolari (MO)
Come MO di partenza per il metodo SCF (e che ci
daranno un’idea qualitativa del legame) usiamo
• approssimazioni più semplici da manipolare delle soluzioni esatte in ξ ed η di +
2H • la stessa dipendenza da φ delle soluzioni
esatte di +2H
Università di Roma “ La Sapienza” mb-9
Dipartimento di Chimica Prof. Guido Gigli
• Cerchiamo ,quindi, una Ψ approssimata usando il metodo variazionale Quale Ψ di prova usare? • per abr grande
Quando l’elettrone è vicino al nucleo a sarà ben descritto dalla autofunzione dello stato
rH es −==Ψ 21
11π
← fondamentale dell’atomo di H
Idem quando è nei pressi di b
per abr piccolo Il sistema tende a diventare lo
ione +He r
He es 221
23 121 −==Ψπ
Dovremmo usare una Z diversa per ogni distanza rab
Z puo’ essere un parametro da ottimizzare a distanza rab fissata
• Poiche’ ad rab grandi l’elettrone risente di uno solo dei due nuclei e’ naturale cercare la Ψ come
bbaa scsc 11 +=Ψ
da determinare variazionalmente
combinazione lineare di orbitali atomici centrati sui due nuclei
Università di Roma “ La Sapienza” mb-10
Dipartimento di Chimica Prof. Guido Gigli
In sintesi stiamo per affrontare la
Versione lineare del Metodo Variazionale ( nella sua forma LCAO )
=Ψprova =Ψ bbaabbaa
iii scscccc 11 +=+=∑ φφφ
• in accordo con il principio variazionale cerchiamo
il minimo (in funzione dei ic ) di
=E =H =ΨΨ
ΨΨ H ∑
∑
ijijji
ijijji
Scc
Hcc
dove il significato dei simboli è il seguente
jiij HH φφ ˆ= jiijS φφ=
• eseguendo le opportune derivate in funzione dei coefficienti e ponendole uguali a zero si ottiene un insieme di equazioni lineari nei coefficienti ic :
- per rab= finito ( nella molecola) Haa<E1s perchè l’elettrone è attratto da entrambi i nuclei
Università di Roma “ La Sapienza” mb-14
Dipartimento di Chimica Prof. Guido Gigli
=−−∇−== bba
abelaab srr
ssHsH 1112
11ˆ12
( ) =−+= ba
abbs1a s1r1s1s1s1Es1
( ) ( )∫−⋅= v1*1
1 drssSE
a
baabs
( ) 012
<+−−= − abrab
abab erSH
• In conclusione possiamo ricavare nneln VEV += con le due soluzioni di −+ EedE
ablegante r
EE 1+= + ab
eantilegant rEE 1+= −
• Notiamo che :
leganteE ed eantilegantE ( −+ EedE ) non sono simmetricamente disposte in energia
( 0>S →< 0abH antilegante più in alto )
a b
ra
carica dovuta alla sovrapposizione fra gli 1s di a e b
β
è un integrale ad- un elettrone - due “centri”
Università di Roma “ La Sapienza” mb-15
Dipartimento di Chimica Prof. Guido Gigli
Università di Roma “ La Sapienza” mb-16
Dipartimento di Chimica Prof. Guido Gigli
• Il risultato energetico complessivo è soltanto
qualitativamente corretto
calc sper re 2.49 2.00 De 0.0648 0.1025 Si può migliorare il risultato usando un altro parametro variazionale come la carica Z* ( vedi mb-9 ) Z*
Re 2.02 2 De 0.0864 1 (per ogni 1s si usa: rab
( ) rZeZs *2/32/1 *1 −−→ π Z* viene ottimizzato per ogni rab ) • Le funzioni d’onda si ottengono sostituendo E+ ed
E- nelle eq. di partenza
( )( )
−===
→
−=Ψ+=Ψ
−−
++
ba
ba
ba
ba
cccc
ssNssN 1
1111
( ) ( ) 2/12/1 12 −−+ += abSN
( ) ( ) 2/12/1 121
abSN
−=−
( )abSfNN =−+ ,
Università di Roma “ La Sapienza” mb-17
Dipartimento di Chimica Prof. Guido Gigli
• l’aspetto delle Ψ± e’ il seguente: • Si puo’ notare che l’addensamento di carica fra i
nuclei e’ maggiore di quanto si otterrebbe con la semplice somma delle densita’ di carica atomiche separate
Somma semplice delle densita’ di
probabilita’
( ) ( )[ ]babaab
ssssS
1121112
1 222 ⋅+++
=Ψ+
( ) ( ) ( )[ ]22 1111212
1baabba
abssSss
SDifferenza +−⋅
+=
( )22 1121
ba ss +=
Università di Roma “ La Sapienza” mb-18
Dipartimento di Chimica Prof. Guido Gigli
Università di Roma “ La Sapienza” mb-19
Dipartimento di Chimica Prof. Guido Gigli
• Questa osservazione sembra attribuire la
formazione del legame essenzialmente alla diminuzione di energia elettronica dovuta alla interazione dell’elettrone con due nuclei anzichè uno solo
IN REALTA’
si devono considerare anche questi fenomeni:
Aumento della repulsione fra i nuclei al diminuire
di abr Aumento dell’esponente negli orbitali atomici (1.24
ad er ) rispetto ad 1 a distanza infinita ( 2 ad 0=abr , 1 ad ∞=abr )
Accumulo di carica vicino ai nuclei (contrazione degli orbitali) e conseguente diminuzione della energia potenziale Diminuzione della componente ⊥ al legame della
energia cinetica (ed aumento della Energia cinetica Totale)
• La questione è complicata, non ancora del tutto
chiarita e, comunque, diversa da molecola a molecola ( lo +
2H potrebbe essere un caso particolare )
• Per lo +2H non è lo spostamento di carica
elettronica nella zona fra i due nuclei che abbassa l’energia ( e stabilizza la molecola ) ma piuttosto la contrazione degli orbitali vicino ai due nuclei che ne viene, di conseguenza, consentita
Università di Roma “ La Sapienza” mb-20
Dipartimento di Chimica Prof. Guido Gigli
Stati eccitati di +2H
• Le opportune funzioni d’onda si possono
“costruire” con il metodo LCAO usando orbitali idrogenoidi degli stati eccitati
Rispettando la simmetria
( )bag ssNs 222 +=σ ( )bau ssNs 222 −=σ
( )zbzag ppNp 222 −=σ ( )zbzau ppNp 222 +=σ
Notare il diverso segno (che può essere convenzionale)
Università di Roma “ La Sapienza” mb-21
Dipartimento di Chimica Prof. Guido Gigli
un’altra classe di funzioni approssimate è quella
degli orbitali π
con termini 1=± me imφ per esempio:
( )xbxaxu ppNp 222 +=π ( )xbxaxg ppNp 222 −=π
• Un modo diverso di vedere queste “combinazioni”
è quello di considerare il problema variazionale come un unico problema nel quale si usa un insieme ( “set” ) di funzioni “base” più ampio
non soltanto
1s o 2s oppure 2p ma
1s, 2s, 2p,ecc.
Notiamo i piani nodali paralleli e perpendicolari all’asse di legame
Università di Roma “ La Sapienza” mb-22
Dipartimento di Chimica Prof. Guido Gigli
• Da due (1s) orbitali atomici, cioè da un “set” di
base minimo ( gli orbitali atomici occupati negli atomi separati) derivavano due orbitali molecolari
ora il numero di orbitali molecolari aumenta MA
Le proprietà di simmetria rendono nulli vari coefficienti in ciascuna combinazione lineare
orbitali atomici con energie molto diverse si mescolano poco
orbitali atomici con scarsa sovrapposizione non contano ai fini del legame
Si possono includere nelle combinazioni lineari i soli orbitali atomici più esterni ( di valenza ) tralasciando quelli più interni (di “core” ) Se ne può avere conferma nelle energie di ionizzazione degli orbitali interni delle molecole che sono pressochè uguali a quelle degli orbitali atomici
a b a
b
per0≅>>− ababaabb SedHHH
si ha:
aabb
abbb HH
HHE−
+≅−
2
aabb
abaa HH
HHE−
−≅+
2
Università di Roma “ La Sapienza” mb-23
Dipartimento di Chimica Prof. Guido Gigli
• Ora, tenendo anche conto delle differenze di energia fra orbitali s, p, d, ecc. in atomi più complessi si possono rappresentare le corrispondenze fra le due situazioni estreme
atomi uniti legame atomi separati gσ (1s)
uσ ( 2pz)
φ φφ
φ φ φ
la simmetria cilindrica si conserva
questi stati
sono CORRELATI
Università di Roma “ La Sapienza” mb-24
Dipartimento di Chimica Prof. Guido Gigli
Struttura Elettronica di Molecole Biatomiche Omonucleari
• Ora passiamo a fare l’ Aufbau degli elettroni
(come per gli atomi polielettronici) rispettando il principio di esclusione di Pauli (e le regole di Hund)
per esempio:
( )22 1sH gσ
( ) ( )2u2
g2 s1s1He σσ nascono delle ambiguità originate dalle intersezioni dello schema di correlazione e, quindi, la sequenza degli orbitali non è univoca
Università di Roma “ La Sapienza” mb-25
Dipartimento di Chimica Prof. Guido Gigli
• Con questo semplice schema di “riempimento” si predicono bene ( qualitativamente ) le proprietà di legame e di spin totale delle molecole biatomiche omonucleari ( ordine del legame = BO “ Bond Order” = (nB-nA)/2 )
Simboli di termine • Come si vede dalla tabella precedente anche per
le molecole si usano simboli per specificare, per ciascun livello energetico, altre caratteristiche
Altre COSTANTI del MOTO
Λ -valore assoluto del momento angolare totale
rispetto all’asse -Λ iiLM λ∑=≡ -E’ solo questa componente che è un “ buon numero quantico ”
infatti - la simmetria assiale del campo elettrico
produce una situazione simile a quella di un atomo in un campo elettrico
- l’accoppiamento è forte ed a stati di moto con diverso LM competono energie molto diverse
- in definitiva è piu’ appropriato classificare gli stati elettronici con LM che con L
S -Spin totale g,u -Parità → utile nelle regole di selezione ( la parità deve cambiare ) +,- -Simmetria ed antisimmetria rispetto ad un piano contenente l’asse di legame ( solo per statib ∑ ) • Non entriamo nel dettaglio di come
da una si arriva ai simboli di configurazione → termine
Università di Roma “ La Sapienza” mb-27
Dipartimento di Chimica Prof. Guido Gigli
Molecole Biatomiche Eteronucleari • La asimmetria dei due atomi ha, qualitativamente,
due conseguenze ➪ Diversa energia degli
orbitali atomici ➪ Distribuzione non uniforme della
densità elettronica
bbaa CC Ψ+Ψ=Ψ 22
ba CC ≠
per esempio, per lo HF
gia’ vista
Il legame diviene parzialmente o
prevalentemente POLARE
Università di Roma “ La Sapienza” mb-28
Dipartimento di Chimica Prof. Guido Gigli
Osservazioni semi-finali
• Tutto quanto abbiamo visto sino ad ora è molto
qualitativo
➪ abbiamo introdotto l’idea di trovare funzioni d’onda molecolari approssimate come combinazioni lineari ( da ottimizzare variazionalmente ) di orbitali atomici centrati sui nuclei degli atomi
LCAO – MO ➪ Per analogia con gli atomi abbiamo disposto
gli elettroni in questi orbitali ➪ Abbiamo descritto brevemente i simboli con i
quali si identificano altre costanti del moto
• Non abbiamo preso in considerazione in modo
esplicito
La repulsione fra gli elettroni
i requisiti di simmetria ed antisimmetria delle Ψ totali
Università di Roma “ La Sapienza” mb-29
Dipartimento di Chimica Prof. Guido Gigli
Molecola H2 • E’ la più semplice con più di 1 elettrone
6 coordinate L’Hamiltoniano elettronico è:
122121
22
21 11111
22ˆ
rrrrrH
bbaael +−−−−∇−∇−=
Lo possiamo riscrivere per mettere in evidenza quelle parti di esso per le quali conosciamo le soluzioni ✴ approccio di tipo MO ( Molecular Orbital )
1222
22
11
21 111
211
2ˆ
rrrrrH
babael +
−−∇−+
−−∇−=
1
2ˆ +HH 2
2ˆ +HH
'ˆˆˆ 0 HHHel +=
( ) ( )2122
0++ Ψ⋅Ψ=Ψ
HH
1ar
12r
abr
2br2ar 1br
a b
1 2
Università di Roma “ La Sapienza” mb-30
Dipartimento di Chimica Prof. Guido Gigli
✴ Approccio di tipo V B ( Valence Bond )
−∇−+
−∇−=
2
22
1
21 1
21
2ˆ
bael rr
H 1212
111rrr ba
+−−
1ˆ HH 2ˆ HH 1221
111rrr ba
+−−
0H
parrebbe che si possa usare indifferentemente ( ) ( )210
bHaH Ψ⋅Ψ=Ψ oppure ( ) ( )210aHbH Ψ⋅Ψ=Ψ
IN REALTA’ nessuna delle due funzioni precedenti è, come deve essere, simmetrica od antisimmetrica e, quindi, si usano le combinazioni:
( ) ( ) ( ) ( )21210aHbHbHaH Ψ⋅Ψ±Ψ⋅Ψ=Ψ
(che tengono conto correttamente della indistinguibilità degli elettroni) • Vediamo il Metodo MO • all’ordine zero gli stati molecolari sono quelli dello
ione +2H
( )bag ssN 11 +⋅= +σ con due elettroni nell’orbitale legante
( ) ( )21 1111 babagggg ssssN +⋅+⋅==⋅=Ψ σσσσ
considerando anche lo Spin
( )βααβσσ −⋅⋅=Ψ gg
o viceversa
Università di Roma “ La Sapienza” mb-31
Dipartimento di Chimica Prof. Guido Gigli
oppure, più dettagliatamente
( ) ( ) ( ) ( ) ( ) ( )[ ]212121 αββασσ −⋅=Ψ ggN ed in forma di Determinante di Slater:
[ ]bbaaabba ssssssssN 11111111 +++= 1 2 1 2 1 2 1 2 i vari termini rappresentano le seguenti situazioni fisiche HH − HH − +− −HH −+ −HH COVALENTEΨ IONICAΨ
• Notiamo che il metodo MO “pesa” in ugual misura le Ψ di tipo covalente e ionica
Università di Roma “ La Sapienza” mb-32
Dipartimento di Chimica Prof. Guido Gigli
• Vediamo il Metodo VB
( ) 01111 Ψ=+=Ψ abbaVB ssssN HH − HH − Il termine ionico è del tutto assente • Un modo diverso di considerare questa Ψ (e che
è il modo con il quale è stata introdotta) è quello di ritenere dominanti nella molecola unicamente quelle “strutture” elettroniche nelle quali gli elettroni sono appaiati (nei vari modi possibili)
La Ψ è una “sovrapposizione” di tutte le strutture “canoniche” Risonanza fra le strutture canoniche
• Anche in questo metodo la energia viene calcolata come valore di aspettazione ( H al completo , VBΨ approssimata )
• In questo caso compaiono soltanto integrali a due centri e due elettroni( ) ( )abbababa e
in accordo con l’Hamiltoniano di ordine zero
Università di Roma “ La Sapienza” mb-33
Dipartimento di Chimica Prof. Guido Gigli
Risultati con i metodi MO e VB
eVE
uarab /
• Si nota che: ➪ Sia VB che MO prevedono uno stato legato
per lo 2H ➪ La curva di potenziale VB dissocia al limite
corretto mentre quella MO dissocia ad energie asssai maggiori
La MOΨ è una sovrapposizione di stati che ad abr grandi “pesano” per metà una situazione +− HH ..... più elevata in energia di quella vera HH .....
➪ Quantitativamente : rispetto ad una ( ) eVveraDe 75.4=
con con s1 “normali” s1 a cariche efficaci VB 3.20 3.78 MO 2.60 3.49
confrontabili80% e 73% della De vera
Università di Roma “ La Sapienza” mb-34
Dipartimento di Chimica Prof. Guido Gigli
Correzioni ai trattamenti VB e MO • Vedendo le cose dal punto di vista della teoria delle
perturbazioni osserviamo che la perturbazione modifica le Ψ di ordine zero “mescolando” altri stati dell’atomo di Idrogeno in aggiunta quello usato ( 1s )
• Si usano, quindi, funzioni miste “polarizzate”
( )rZrZ ezeNs '''1 −− +→ λ del “tipo” 1s 2p parametro variazionale • Si usa, anche, una forma mista VB – MO
IONICACOVALENTE Ψ+Ψ=Ψ λ
parametro variazionale
➪ Di fatto, nel linguaggio del metodo VB, si
introduce una RISONANZA ionico-covalente
➪ In questo modo si alleviano le conseguenze del difetto principale del metodo VB:
sovrastimare la correlazione elettronica ( non considera strutture con entrambi
gli elettroni sullo stesso atomo )
In sostanza si usa un orbitale IBRIDO alla ricerca di una migliore sovrapposizione
Università di Roma “ La Sapienza” mb-35
Dipartimento di Chimica Prof. Guido Gigli
• Si espande la Base Variazionale usata con altri
determinanti di Slater che includono configurazioni eccitate ( )uu σσ con la medesima simmetria
➪ Nel linguaggio del metodo MO si introduce la
Interazione di Configurazione ( CI )
➪ In questo modo si corregge il difetto principale del metodo MO Sottostima della correlazione elettronica
(strutture con entrambi gli elettroni sullo stesso atomo hanno peso pari a quelle con un elettrone su ciascun atomo)
( ) ( ) ( )covalenteionica Ψ−+Ψ+= λλ 1)(1 • Si usano funzioni con dipendenza esplicita da 121 r
1933 James e Coolidge Ψ con 13 termini 724De .=
1986 Kolos et Wolniewicz Ψ con 279 termini picospettroscoDD 00 =
( entro 0.1cm-1 ! )
Università di Roma “ La Sapienza” mb-36
Dipartimento di Chimica Prof. Guido Gigli
Un osservazione sul problema della Correlazione • Il problema ha due aspetti
➪ “Spaziale”
L’approssimazione introdotta può trascurare la distorsione “locale” della distribuzione di elettroni. Per esempio nel metodo HF un orbitale dovrebbe essere distorto nelle zone in cui è vicino ad un altro elettrone. Viceversa tutto l’orbitale viene distorto in un modo medio � la prob. di trovare due elettroni nello stesso punto non è nulla
➪ di “Spin”
Quando si scrive una Ψ in modo tale che sia antisimmetrica compaiono termini “di scambio” . In sostanza, allora, mentre il singolo elettrone risente soltanto di un campo coulombiano medio di tutti gli altri, è sottoposto ad un effetto istantaneo a causa degli elettroni con il medesimo Spin � la prob.di trovare due elettroni contemporaneamente nello stesso punto dello spazio e con il medesimo Spin è nulla
Università di Roma “ La Sapienza” ml-1
Dipartimento di Chimica Prof. Guido Gigli
LCAO – MO SCF per molecole • E’ la base dei calcoli “ab-initio” della struttura
elettronica di molecole • Hamiltoniano
∑ ∑ ∑= = <
+−∇−=
n
i
M
ji iji
iel rr
ZH1 1
2 12
ˆα α
α
• Assumiamo che la Ψ sia esprimibile come un
Determinante di Slater di MO tutti occupati ( 2 elettroni per orbitale )
RHF ( Restricted Hartree Fock )
( ) ( ) ( ) ( ) ( ) ( )
( ) ( ) ( ) ( )NNNN
N
n
n
βφαφ
βφβφαφ
21
211 111111
LL
MMMM
MMMM
L
=Ψ
• Il metodo prosegue in modo essenzialmente
identico a quello visto per gli atomi • La HFE viene valutata variazionalmente
Ψ+Ψ= nnelHF VHE ˆ
cioè si cercano gli orbitali iφ che minimizzano
n elettroniM nuclei
n elettroni N=n/2 orbitali
Università di Roma “ La Sapienza” ml-2
Dipartimento di Chimica Prof. Guido Gigli
• Si trova che i singoli orbitali soddisfano la
equazione kkkkF φεφ =ˆ
( ) ( )[ ]∑ ∑ −+−∇
−=α α
α
jjj
k
kk kKkJ
rZ
F 22
ˆ2
N.B.➪ Già a questo livello il problema è tipicamente
non lineare e richiede la procedura SCF ( mediante iterazioni )
➪ Il problema è già stato molto semplificato • Seguendo Roothan ciascuna φ spaziale ( ciascun
MO ) viene espressa come LCAO
∑∑ ==ν
ννν
ννφ bcAOc kkk
N.B. ➪ Questa “espansione” è di necessità limitata
ad un numero finito di AO e, quindi, imperfetta � Si introduce qui quello che viene chiamato
ERRORE di TRONCAMENTO
Potenziale medio dovuto agli altri elettroni
Università di Roma “ La Sapienza” ml-3
Dipartimento di Chimica Prof. Guido Gigli
• Sostituendo la LCAO nelle equazioni di Fock si
ottiene una equazione secolare
per ciascuna k ( ) 0=−∑ νν
µνµν ε cSF
νµ bb=
νµ bFb ˆ=
• La soluzione si trova per
0=− SF ε MATRICI
vi compaiono integrali monoelettronici e bielettronici
• La difficoltà del metodo è nell’ordine di grandezza
del numero di questi integrali Per quelli a due elettroni ( )4NO N=numero di
AO usati nel LCAO
( )2NO a causa della entità di alcuni
vi sono
due esigenze contrastanti ➭ elevato numero di AO usati (tante Funzioni
di Base ) ➭ piccolo numero di integrali
Da risolvere ITERATIVAMENTE F dipende dagli orbitali che, a loro volta, dipendono dai coefficienti
come pergli
atomi
Università di Roma “ La Sapienza” ml-4
Dipartimento di Chimica Prof. Guido Gigli
Lo schema di larga massima della procedura è 1. si fissa una geometria molecolare 2. si scelgono le “funzioni di base” ( gli AO ) 3. si calcolano tutti gli integrali che la formulazione
HF prevede ( di “core”, di sovrapposizione, di scambio )
4 si ipotizzano i coefficienti νkC delle LCAO 5 si calcolano le matrici F 6 si risolve il problema degli autovalori ed
autovettori ( Energie e coefficienti ) 7 si ritorna al 5 o si termina Funzioni di base ( “Set” di base ) • In linea di principio si possono usare orbitali
idrogenoidi ed anche orbitali HF MA
la loro forma analitica rende difficile il calcolo degli integrali ( in particolare la presenza di “nodi” )
• Si usano prevalentemente
STO ( Slater Type Orbitals ) � in calcoli semiempirici
❒ anche questi poco usati nei calcoli HF per le difficoltà di integrazione
Università di Roma “ La Sapienza” ml-5
Dipartimento di Chimica Prof. Guido Gigli
GO – GTO Gaussian Orbitals
( )φθα ,,2
mlrcba Yezyxg −=
coordinate cartesiane α parametro con origine nell’atomo considerato a, b, c interi
un orbitale atomico realistico può essere “costruito” con varie Gaussiane
• Sono stati proposti molti schemi di combinazioni di funzioni gaussiane semplici o piu’ o meno composte per tenere conto di esigenze contradditorie
➜ rendere l’insieme delle funzioni di base quanto più possibile esteso e flessibile
➜ mantenere il numero e la difficoltà dei calcoli da eseguire entro limiti accettabili
• c’è una moltitudine di “set” di base ciascuno
contraddistinto da una sigla che ne ricorda le caratteristiche; per esempio
6-311 G ✳✳ 6 Gaussiane cosiddette primitive sono usate per
rappresentare gli orbitali interni 311 – identifica il numero e tipo di gaussiane
usate per gli orbitali ✳ ✳ - Il set di base contiene funzioni particolari(di
polarizzazione): per atomi diversi da H ( primo asterisco ) per atomi di H ( secondo asterisco )
Gli integrali sono più semplici
Università di Roma “ La Sapienza” ml-6
Dipartimento di Chimica Prof. Guido Gigli
Esempio di calcoli HF per H2O • Set di Base 6-311 G ✴ ✴ ( in totale 30 funzioni )
- per ogni H 3 funzioni s 3 funzioni p ( polarizzazione )
- per ogni 0 1 funzione dei gusci interni ( “core”) 3 funzioni s di valenza 3 insiemi ( x3 ) funzioni p 1 insieme ( x5 ) di funzioni d
MO 1 2 3 4 5 6 7
E -20.541 -1.3492 -.71721 -.57287 -.50066 .15259 .21857
Funzioni H s -.00021 .09656 .15126 -.08748 -.03413 -.02364 H s -.00008 .08131 .21224 -.14604 .06832 .13008 H s -.00010 -.00267 .05449 -.02727 -.84441 -1.58014 H px .00007 -.02386 -.01870 .02973 -.00513 -.00885 H py .03154 H pz .00001 -.01441 -.02567 -.00956 .00381 -.00819 O s -.55143 -.11336 -.03815 -.03350 O s -.47168 -.18936 -.06487 -.05463 O px .22740 .12024 O py .29169 O pz -.00178 .03804 -.25567 .07159 O s -.00557 .53789 .19649 .10269 O px .34889 .12747 O py .43668 O pz .00062 .06315 -.37815 .11396 O s .00046 .37189 .33491 .85269 O px .21181 .49547 O py .46589 O pz -.00009 .02078 -.33996 .19994 O dz2 .00002 .00276 -.01699 .00456 O dy2-z2 .00012 .00804 -.00506 .00367 O dxy O dxz .02933 .00553 O dyz .01714 H s -.00021 .09656 -.15126 -.08748 -.03413 .02364 H s -.00008 .08131 -.21224 -.14604 .06832 -.13008 H s -.00010 -.00267 -.05449 -.02727 -.84441 1.58014 H px -.00007 .02386 -.01870 -.02973 .00513 -.00885 H py .03154 H pz .00001 -.01441 .02567 -.00956 .00381 .00819
Università di Roma “ La Sapienza” ml-7
Dipartimento di Chimica Prof. Guido Gigli
3a1
5 6 7 1b1 4a1 2b2
Università di Roma “ La Sapienza” ml-8
Dipartimento di Chimica Prof. Guido Gigli
Notiamo: orbitale 1 è localizzato sull’ossigeno ( i coefficienti
degli atomi di H sono trascurabili )
• non è coinvolto nel legame • anche se una parte notevole della
energia totale è dovuta a questo contributo il suo livello energetico è molto più in basso di quello degli altri orbitali
orbitale 2 pur estendendosi fino ai nuclei di H i
coefficienti relativi sono piccoli • c’è un “piano” nodale fra O ed H • l’energia è piuttosto diversa da quella
degli atomi di H isolati( - 0.5 )
Debolmente coinvolto nel legame
orbitali 3 e 4 i coefficienti, sia per O che H, sono
grandi e non ci sono superfici nodali fra gli atomi
• hanno energia confrontabile con gli orbitali 1 s
• il 3 è simile al px dell’ossigeno il 4 è simile al pz dell’ossigeno
Forniscono il massimo contributo al legame
Università di Roma “ La Sapienza” ml-9
Dipartimento di Chimica Prof. Guido Gigli
orbitale 5 simile al py dell’ossigeno
scarso “contenuto” di H ( si può mescolare soltanto con le funzioni di polarizzazione degli Idrogeni )
E’ sostanzialmente un “ lone pair” ( è occupato ) essendo l’ultimo orbitale molecolare occupato viene chiamato HOMO
orbitale 6 Energia positiva - antilegante
è il primo non occupato LUMO
carattere prevalentemente 3 s
orbitale 7 Energia positiva - antilegante ha piani nodali fra 0 ed H
• Complessivamente la sequenza in energia degli
orbitali molecolari che abbiamo trovato è simile a quella ricavabile da considerazioni più qualitative sulla formazione degli stessi
Università di Roma “ La Sapienza” ml-10
Dipartimento di Chimica Prof. Guido Gigli
O H2O H • Gli spettri di Fotoelettroni confermano la struttura
elettronica emersa dal calcolo LCAO-MO-SCF
• Per quanto riguarda la geometria di equilibrio della
molecola la si trova cercando il minimo della energia elettronica al variare della geometria stessa
Si esplora la superficie di Potenziale ( approssimazione di Born-Oppenheimer )
Anche per le molecole
poliatomiche si usa la notazione
σ e π ma il significato è ora connesso alla
simmetria e non al momento angolare
Gli MO sono identificati con simboli che ne chiariscono la simmetria ( nel
gruppo C2V )
Da quali combinazioni di AO
vengono forniti
Università di Roma “ La Sapienza” ml-11
Dipartimento di Chimica Prof. Guido Gigli
Affidabilità dei calcoli HF • Dipende dalla “base” usata • Per il II periodo, tentando una generalizzazione :
♦ lunghezze di legame piuttosto accurate ( entro 0.05 A )
♦ Frequenze di vibrazione maggiori di quanto osservato sperimentalmente del 10 ÷ 15% E’ consueto applicare un fattore empirico, spesso pari a ∼ 0.90, ai valori calcolati
♦ Energie di Dissociazione spesso troppo piccole ( decine di % )
Orbitali Molecolari localizzati • La descrizione della struttura elettronica con il
metodo MO non mette in primo piano la formazione dei legami chimici fra atomo ed atomo
Per la sua natura un MO ( anche legante ) è diffuso su tutta la molecola
• Un calcolo MO fatto per una molecola come CH3OH ci fornisce forme degli orbitali molecolari piuttosto diverse da quelle della H2O
Università di Roma “ La Sapienza” ml-12
Dipartimento di Chimica Prof. Guido Gigli
Università di Roma “ La Sapienza” ml-13
Dipartimento di Chimica Prof. Guido Gigli
PERALTRO • Evidenze di tipo chimico e spettroscopico indicano
che per esempio il legame OH conserva una sua “identità” nelle varie molecole in cui è presente
In generale, per esempio, la lunghezza di un legame è ben “trasferibile” da un edificio molecolare ad un altro
• In realtà è possibile formulare il metodo in modo
diverso per mettere in luce più chiaramente una distribuzione di densità elettronica localizzata lungo i legami chimici
INFATTI
Ce lo consente la proprietà dei determinanti di essere invarianti alla combinazione di righe o colonne Ad una descrizione altrettanto valida della molecola di H2O si arriva combinando linearmente gli orbitali delocalizzati che abbiamo trovato Per esempio gli orbitali 2 e 3 possono essere “combinati” come 2 - 3 e 2 + 3
a1 - b2 a1 + b2
Università di Roma “ La Sapienza” ml-14
Dipartimento di Chimica Prof. Guido Gigli
SCHEMATICAMENTE 2a1 - 1b2 2a2+1b2 • In modo analogo si possono ottenere altri orbitali
non canonici che meglio corrispondono alla visione chimica consueta ( ma non rispettano la simmetria e non riproducono la energia )
Dai 3a1 e 1b1 si possono ricavare degli orbitali localizzati che riproducono la struttura elettronica a “ Doppietti Isolati “
• In verità talora può interessare mettere in evidenza
la localizzazione del legame, talaltra no.
Legame Localizzato Delocalizzato Energia del legame Spettro elettronico lunghezza “ Fotoionizzazione costante di forza Spettro IR
Università di Roma “ La Sapienza” ml-15
Dipartimento di Chimica Prof. Guido Gigli
Metodo VB ( Molecole Poliatomiche ) • Proprio dal punto di vista “chimico” dell’edificio
molecolare come insieme di orbitali/legami localizzati prende le mosse il metodo VB
• Il metodo prende in considerazione, sin dall’inizio i doppietti di elettroni ( alla LEWIS )
➭ ci sono vari metodi per accoppiare gli elettroni in
tutti i modi possibili ➭ ciascun modo è una struttura elettronica ➭ per ciascuna struttura si scrive una ψ
polielettronica alla Heitler-London • La Ψ totale è una combinazione lineare (
sovrapposizione ) delle varie strutture
∑ Φ=Ψi
iiC
➭ i coefficienti Ci vengono determinati
variazionalmente ➭ la Ψ complessiva è denominata
Ibrido di Risonanza tra le varie strutture
Università di Roma “ La Sapienza” ml-16
Dipartimento di Chimica Prof. Guido Gigli
Per esempio per la H2O • Si considerano come possibili partecipanti ai
legami e, quindi, da considerare nello scrivere le φi i soli elettroni spaiati ( ) ( ) ( )sHsHppO zy 11,
• le strutture covalenti ( CANONICHE ) da considerare sono:
A O B O C O H H H H H H 2py 2Pz 2py 2Pz 2py 2Pz 1s 1s 1s 1s 1s 1s
quelle Ioniche sono
D 1-O E 1-O F 2-O H H+ H+ H H+ H+
G 1O+ H 1O+ I 2O+
H 1-H 1-H H 1-H 1-H
[ ] FFEDEDBBAATOT cccc Ψ+Ψ+Ψ+Ψ+Ψ=Ψ ,
FEDBA ccccc ,,,, sono ricavati variazionalmente otteniamo un Determinante Secolare tante ψtot ed Etot quante sono le strutture incluse
nel processo variazionale l’energia totale del fondamentale si abbassa in
dipendenza del numero di strutture risonanti Energia di Risonanza
è una combinazione delle prime e, quindi, da non considerare
tutte trascurabili
Università di Roma “ La Sapienza” ml-17
Dipartimento di Chimica Prof. Guido Gigli
Da notare:
Nel metodo VB non c’è un orbitale molecolare
funzione monoelettronica estesa a tutta la molecola
vi sono soltanto Determinanti Polielettronici • Per massimizzare la localizzazione elettronica e la
equivalenza dei legami il metodo VB ( Pauling ) ha introdotto il concetto di
IBRIDIZZAZIONE
per esempio: alla consueta ibridizzazione sp3 del
carbonio si arriva ipotizzando che la energia persa nella promozione di due elettroni ( 3P0 � sp3 ) del carbonio sia più che guadagnata nella successiva formazione di legami
sp3 ( )zyx pppsN 22221 ++++=χ
8.26 eV ( )zyx pppsN 22222 +−−+=χ 3P0 25.31 eV ( )zyx pppsN 22223 −−++=χ
C ( )zyx pppsN 22224 −+−+=χ CH4
( )
...............................
...............................1111 χφ bsaN +=
( )4414 χφ bsaN +=
Università di Roma “ La Sapienza” ml-18
Dipartimento di Chimica Prof. Guido Gigli
• Il concetto di RISONANZA ( non nel tempo ) è un
aspetto fondamentale del metodo VB
consente la “flessibilità” variazionale • Da notare anche che:
Ibridizzazione, Risonanza, Energia di Risonanza non sono reali fenomeni fisici ma conseguenza delle approssimazioni introdotte così come gli orbitali, in sé per sé, non esistono e la Interazione di Configurazioni non è reale
confronto MO – VB
MO è più facile da usare perchè gli elettroni sono in orbitali ortogonali
VB è più difficile perchè il numero di strutture
covalenti e ioniche da considerare può essere molto grande (sviluppi recenti riducono questo difetto)
MO “correla” male con gli stati a distanza grande
fra gli atomi
è particolarmente inefficiente nel predire le energie di dissociazione
Università di Roma “ La Sapienza” ml-19
Dipartimento di Chimica Prof. Guido Gigli
Interazione di Configurazioni • Ne abbiamo visto un esempio elementare per lo H2 • I metodi CI (Configurations Interaction) tengono
conto della correlazione scrivendo la funzione d’onda come combinazione lineare di molti Determinanti da ottimizzare variazionalmente
∑=Ψi
ii DC
• La ottimizzazione variazionale per i Ci conduce ad
un Det. Secolare
0=− SEH • Il metodo, affinchè si realizzi un reale
miglioramento, richiede parecchie configurazioni ed il Det. diviene estremamente grande (nell’ordine dei milioni)
un elettrone promosso ad orbitale virtuale
due elettroni promossi ad
un orbitale virtuale tutti promossi FCI
SDCI
Università di Roma “ La Sapienza” ml-20
Dipartimento di Chimica Prof. Guido Gigli
Metodi DFT • Sono basati su alcuni teoremi dovuti a Hohenberg
e Kohn; e Kohn e Sham ( 1964 – 1965 ) • Il punto principale è stato la dimostrazione che
qualunque proprieta’ molecolare dello stato fondamentale ( e tra queste l’Energia ) può essere fornita con esattezza da un Funzionale della densità elettronica ρ della molecola
[ ]ρE
Funzione è una regola che associa un numero
ad ogni valore della variabile X ( ) 23 2 += xxf
Funzionale è una regola che associa un numero
ad una funzione
[ ] ∫∞+
∞−== τdfffffF *
[ ] ΨΨ==Ψ HEE ˆ
• Il sapere che nota ρ si può valutare E è una
semplificazione nel numero delle variabili (ψ → E )
rimangono i problemi di: come si calcola ρ
come si calcola E da ρ
( )xf
F[ ]f
Università di Roma “ La Sapienza” ml-21
Dipartimento di Chimica Prof. Guido Gigli
• Kohn e Sham hanno fornito, in linea di principio,
una soluzione a questi quesiti
scnnen EVVE +++Τ=
[ ]ρscsc EE =
2∑ Ψ=i
iρ
iΨ sono ottenute per soluzione di una equazione
simile a quella HF ricavata dalla minimizzazione variazionale della [ ]ρE
Ψ=Ψ KSKSF ε
( ) ( )∑ ∑ ++−∇−=α α
α
JscJiKS VJ
rZ
F 1211
1
2
ρ∂∂= sc
scEV
• Non ci sono espressioni esatte per Esc e ciò ha
dato origine a vari metodi in cui la Esc dipende da parametri empiricamente adattati a riprodurre le proprietà di classi di molecole
E di Correlazione- -Scambio
Sostituisce gli operatori di scambio dell’op. di Fock
Università di Roma “ La Sapienza” ml-22
Dipartimento di Chimica Prof. Guido Gigli
Per esempio: LDA (Local Density Approximation )
[ ] ∫ ∫
−== τραπ
τρερρ ddE scLDASC
3453
89
B3LYP il metodo attualmente più usato nel quale
sono inserite correzioni che tengono conto del Gradiente della ρ
• In definitiva la procedura di un calcolo DFT ( per
esempio LDA ) prevede: Stima della ρ ( per es. da un calcolo HF od altro
metodo semiempirico ) Calcolo di [ ]ρscE ( per es. con espressioni esplicite
come per il metodo LDA ) Soluzione della eq. di Kohn-Sham
Ψ=Ψ KSKSF ε
Calcolo della nuova ρ con le nuove ψ
Calcolo di un nuovo [ ]ρ
ρ∂
∂= scsc
EV
Si cicla iterativamente ( alla SCF )
parametro Energia di Correlazione e Scambio di elettroni in una scatola ( gas di elettroni )
Università di Roma “ La Sapienza” ml-23
Dipartimento di Chimica Prof. Guido Gigli
Esempio Conclusivo • Come esempio dei risultati ottenibili con i metodi
moderni vediamo le curve di energia potenziale della molecola H2
• L’ottimo accordo del metodo B3LYP è anche
dovuto al fatto che il funzionale di correlazione e scambio è parametrizzato su molte molecole tra cui la stessa molecola H2
Università di Roma “ La Sapienza” ml-24
Dipartimento di Chimica Prof. Guido Gigli
METODI SEMIEMPIRICI • Nei metodi ( ab-initio ) visti sino ad ora si usa:
H esatto Ψ approssimato
• Nei metodi semiempirici si usa: H approssimato
Metodo di Hϋckel • Si prendono in considerazione soltanto gli elettroni
π in molecole:
Planari • Lo “scheletro” di legami σ è fisso e non interagisce
con i π • La repulsione elettrone-elettrone è trascurata
∑=i
itot HH ˆˆ
aromatiche eterocicliche
Università di Roma “ La Sapienza” ml-25
Dipartimento di Chimica Prof. Guido Gigli
• Ogni MO è scritto come LCAO di orbitali “p”
degli atomi di Carbonio
µµ
µφ pCii ∑=
iii EH φφ =ˆ
si ottengono le solite eq. secolari
0=− SEH
• Si fanno alcune drastiche approssimazioni
== νµµν pHpH ˆ
µννµµν δ== ppS ETILENE
22222121
12121111
SEHSEHSEHSEH
−−−−
➜
con β
α Ex −= si ha ➜ = x2 – 1 = 0
E = α ± β x = ± 1
νµ
νµβνµα
e di nicombinazio altre le tutte per 0
legatie per per =
α e β - non sono calcolati- sono parametri
EE
−−
αββα
x 1 1 x
Università di Roma “ La Sapienza” ml-26
Dipartimento di Chimica Prof. Guido Gigli
02
1 =
−
−CC
EE
αββα
per E+ = α + β
C1=C2 e normalizzando 02
1 =
−
−CC
ββββ
( )2121 pp +=+φ
analogamente
( )2121 pp −=−φ ( )βα += 2totE
β2=−=∆ +− EEE
β ≈ 30 ÷ 80 kJ/mole Da altre molecole o
proprietà si ha la variabilità è un test dlla teoria BUTADIENE • Indipendentemente da CIS o TRANS il Det. Secolare
( Det. Topologico ) è:
xx
xx
EE
EE
100110011001
000
000
=
−−
−−
αββαβ
βαββα
Si stima β dalla transizione dell’etilene β ≈ 75 kJ/mole
α − β α β α + β
Università di Roma “ La Sapienza” ml-27
Dipartimento di Chimica Prof. Guido Gigli
Sviluppando:
xx
xxx
xxx
xx
xx
x1
111
011
11
1
1010
0111
101101
⋅⋅−
⋅−
⋅⋅=⋅−⋅
( ) ( ) 013111 24222 =+−=−−⋅−−= xxxxxxx
±=±=±+=−±+=
618.0618.1
253
24932
xx
x
Da notare: la Energia cresce con il numero di nodi Etot = 4 α +4.48 β 2 Etot (Etilene ) =4α +4β
Due legami Due legami Delocalizzati ∆ E =0.48 β Energia di Delocalizzazione
Il Det. Secolare ( 6 x 6 ) è quasi uguale agli altri
0
100011100001100001100001110001
=
xx
xx
xx
Nota bene α - 2 β α - β --------------------- α ↑ ↓ ↑ ↓ α +β ↑ ↓ α + 2 β E di Delocalizzazione = Etot –3 Etot (Etilene )
= 2 ( α+2β )+4 ( α+β ) – 3 ( 2α+2β )= = 2β
N.B.
la eq. caratteristica è x6-6x4+9x2-4 =0
Università di Roma “ La Sapienza” ml-29
Dipartimento di Chimica Prof. Guido Gigli
• Si può osservare che l’uso della simmetria
(mediante la teoria dei gruppi) avrebbe consentito di semplificare il problema della risoluzione del Determinante costruendo i cosiddetti
SALC (Symmetry Adapted Linear Combinations)
Per esempio, per il Butadiene, usando il piano di simmetria
2222 CHCHCHCH =−= le ψ di LCAO sarebbero state costruite con φi :
( ) ( )( ) ( )414323
322411
2121
2121
pppp
pppp
−=−=
+=+=
φφφφ
ed il Det. Secolare sarebbe stato:
11001000011001
−
+
xx
xx
• In effetti quando c’è un problema che genera un
Det. secolare lo sforzo di soluzione della eq. di Schroedinger può essere considerevolmente ridotto usando Funzioni di Base coerenti con la simmetria della molecola
Funzioni di Base
Il Det. viene Fattorizzato in Due Det. di ordine inferiore
Università di Roma “ La Sapienza” ml-30
Dipartimento di Chimica Prof. Guido Gigli
• Il Metodo di Hückel fornisce altre proprietà 1. Densità di carica π su ciascun atomo
2µµ i
ii Cnq ∑=
n di elettroni nello MO iesimo 2. Ordine di legame π ( fra µ e ν )
∑ ⋅=i
iii CCnp νµµν
Per l’Etilene
12
12
1212 =
=p
Per il Butadiene ( ) ( )( ) ( )( )
simmetria per2334
23
12
45.037.037.0260.060.0289.037.060.0260.037.02
pppp
==−×+×=
=×+×=
i legami terminali del Butadiene hanno maggior carattere di legame π (in accordo con le lunghezze di legame)
3. Indice di Valenza libera ( Indice della capacità di
formare altri legami π )
∑∑∑ −=−
=ν
µνν
µνν
µνµ pppF 3max
Per il Butadiene
39.084.0 3241 ==== FFFF
Università di Roma “ La Sapienza” ml-31
Dipartimento di Chimica Prof. Guido Gigli
Metodo di Hückel Esteso • Si considerano sia gli elettroni π che i σ con un set
di base minimo ( di valenza ) tipicamente STO • La rep. elettronica è trascurata • Si arriva sempre ad una eq. Secolare 0=− SEH
• Differisce dal metodo di Hückel per la parametrizzazione di H ed S