1 Termodinamica delle soluzioni di polimeri La soluzione ideale e l’ entropia di mescolamento Nel miscelare omogeneamente due sostanze nello stato liquido si ha generalmente una variazione >0 di entropia, ∆S m . L’ entropia è una funzione di stato legata al numero di stati microscopici del sistema. Ad una data T e P oppure ad una data T e V, maggiore è il numero di stati microscopici (o microstati) in cui si può realizzare un sistema, maggiore e’ l’ entropia del sistema. Tale numero è solitamente destinato ad aumentare quando lo spazio venga riempito da molecole di due tipi diversi invece che di un unico tipo. Invece la variazione di entalpia alla miscelazione, ∆H m , può sia corrispondere ad un valore positivo (opponendosi al mescolamento) che ad un valore negativo (promuovendo il mescolamento). Nel caso in cui il calore di mescolamento sia nullo, la soluzione è detta ideale e l’ energia libera di mescolamento, ∆F m , ≡ -T∆S m . Nel caso di soluzioni semplici di molecole piccole valutiamo l’ entropia di mescolamento, assumendo per semplicità: i) Che le molecole delle 2 sostanze abbiano all’ incirca le medesime dimensioni; ii) Che si possano considerare disposte sui nodi di un reticolo; iii) Che siano distribuite in modo completamente casuale. Sia N A il numero di molecole della specie A e N B il numero di molecole della specie B. Sia N il numero totale di siti, N=N A + N B . ∆S m è legato al numero distinto di modi di riempire il reticolo con tali molecole, Ω ΑΒ , a partire da A puro e B puro.
Transcript
1
Termodinamica delle soluzioni di polimeriLa soluzione ideale e l’ entropia di mescolamento
Nel miscelare omogeneamente due sostanze nello stato liquido si ha generalmente una variazione >0 di entropia, ∆Sm. L’entropia è una funzione di stato legata al numero di stati microscopici del sistema. Ad una data T e P oppure ad una data T e V, maggiore è il numero di stati microscopici (o microstati) in cui si può realizzare un sistema, maggiore e’ l’entropia del sistema. Tale numero è solitamente destinato ad aumentare quando lo spazio venga riempito da molecole di due tipi diversi invece che di un unico tipo.
Invece la variazione di entalpia alla miscelazione, ∆Hm, può sia corrispondere ad un valore positivo (opponendosi al mescolamento) che ad un valore negativo (promuovendo il mescolamento). Nel caso in cui il calore di mescolamento sia nullo, la soluzione è detta ideale e l’energia libera di mescolamento, ∆Fm, ≡ -T∆Sm. Nel caso di soluzioni semplici di molecole piccole valutiamo l’ entropia di mescolamento, assumendo per semplicità:i) Che le molecole delle 2 sostanze abbiano all’ incirca le medesime dimensioni;ii) Che si possano considerare disposte sui nodi di un reticolo;iii) Che siano distribuite in modo completamente casuale.Sia NA il numero di molecole della specie A e NB il numero di molecole della specie B. Sia N il numero totale di siti, N=NA
+NB. ∆Sm è legato al numero distinto di modi di riempire il reticolo con tali molecole, ΩΑΒ, a partire da A puro e B puro.
2
Naturalmente, le situazioni di partenza, ovvero il sistema di NA molecole di A su NA siti (A puro) e di NB molecole di B su NB siti (B puro) non puo’ che realizzarsi in unico modo, (ΩΑ=ΩΒ=1) per cui l’ entropia di mescolamento vale: ∆Sm= k lnΩΑΒ − k lnΩΑ - k lnΩΒ = k lnΩΑΒ, con k la cost. di Boltzmann. Per valutare ΩΑΒ, disponiamo le molecole sul reticolo una per volta, ad es. prima le molec. A e poi quelle B, e valutiamo il numero distinto di modi di accomodare dette molecole sul reticolo.Alla prima molecola di specie A sono disponibili N siti. Ciascuna di tali situazioni (N) lascia N-1 siti liberi disponibili per l’ occupazione di un sito reticolare da parte della seconda molecola, per cui il numero di modi di collocare due molecole su N siti risulta N(N –1); per tre molecole N(N –1)(N –2); …per NA molecole N(N –1)(N –2)…(N –NA+1)…per per NA molecole A e la prima di tipo B N(N –1)(N –2)…(N –NA+1) (N –NA)…per NA molec. A e NB molec. B: N(N –1)(N –2)…(N –NA+1)(N –NA)(N –NA-1)…(N –NA- NB-2)1
= N!Ma N! non corrisponde al numero distinto di modi di riempire il reticolo con le due specie di molec. in questione. Infatti, per ciascuna di tali configurazioni è possibile scambiare la posizione delle NA molecole tra di loro (non intaccando la posizione delle molecole B) e lo stesso per le molecole B, (non intaccando la posiz. delle molecole A), senza tuttavia ottenere configurazioni microscopiche del sistema (o microstati) distinguibili tra loro, in quanto le molecole di A sono tra loro indistinguibili e cosi’ anche quelle di B. Per questa ragione il numero distinto di modi di realizzare il sistema in questione, ΩΑΒ, risulta: ΩΑΒ = N!/(NA! NB!) ove al denominatore si tiene conto del fatto che le molecole A sono indistinguibili tra loro e lo stesso per quelle B.
L’ entropia di mescolamento, ∆Sm= k lnΩAB risulta quindi: [ ]!ln!ln!ln BAm NNNkS −−=∆
MMMM −= ln!lnUtilizzando l’ approssimazione di Stirling, valida per M>>1:si ottiene:
[ ] [ ][ ]BBAA
BBAABBBAAAm
xnxnRNNNNNNkNNNNNNNNNkS
lnlnlnlnlnlnln
+−=+−=+−+−−=∆
ove R è la costante dei gas, xA e xB le frazioni molari di A e B e nA e nB le corrispondenti moli (N=NAvn, NAv il numero di Avogadro). Tale quantità e sempre >0, dal momento che xA e xB sono <1. Tale eq. per ∆Smè valida solo se i vari microstati sono tutti equiprobabili. Ciò si verifica nel caso in cui l’ energia di interazione tra le varie specie in soluzione sono tutte uguali per cui le interazioni tra molecole tutte di tipo A sono equiv. a quelle tra molec. tutte di tipo B, e a quelle tra specie diverse. Tale situazione corrisponde ad un’ entalpia di mescolamento nulla, per cui l’energia libera di mescolamento risulta:
Da ciò si ricava che l’energia libera di mescolamento tra due specie per una soluzione ideale èsempre una quantità <0, indicando che il mescolamento è sempre favorito. Si noti che detto vo il volume di una singola cella reticolare, il volume totale è V=N vo, mentre le frazioni in volume occupate dalle molecole A e B (φΑ e φΒ, rispettivamente) risultano φΑ = N Avo / N vo=xA e φβ = NBvo / N vo=xB, per cui l’ entropia di mescolamento può essere anche scritta in termini di detti parametri:
[ ]BBAAmm xnxnRTSTG lnln +=∆−=∆
[ ] [ ]BBAABBAAm kNNNkS φφ+φφ−=φ+φ−=∆ lnlnlnln 3
4
Passiamo ora ad un altro tipo di soluzione, detta soluzione regolare. Per tale soluzione le ipotesi i-iii viste prime continuano ad essere valide, ma si ammette che l’ energia di mescolamento possa essere diversa da zero. Si noti che l’ ipotesi iii) che il mescolamento tra le varie unità sia casuale implica che le interazioni tra le varie specie, favorevoli o sfavorevoli che siano, sono sufficientemente piccole da non influenzare il loro mescolamento casuale. In tale teoria l’ entropia di mescolamento corrisponde a quella delle soluzioni ideali. La teoria delle soluzioni regolari o di Hildebrand prevede che l’energia di mesc. sia espressa in termini di 3 parametri di interazione, uAA, uBB e uAB tra siti adiacenti occupati da due specie.Si immagina che l’energia media d’interazione di una molecola di specie A (UA) che occupi un certo sito reticolare con una seconda molecola che occupa un sito reticolare adiacente possa essere calcolata nell’ ipotesi di campo medio (mean field). La probabilità che questo sito I vicino sia occupato una molec. di specie A viene assunta in base a tale ipotesi pari a φA(ignorando l’ effetto delle interazioni su tale probabilità). La probabilità che tale sito sia occupato da una molecola di specie B risulta invece pari a 1- φA= φB. Pertanto l’ energia media di interazione di coppia di una molecola di tipo A con una molecola collocata in un sito I vicino, UA, è la somma delle energie di interazione possibili a seconda della natura della molecola collocata nel sito I vicino, oppurtunamente pesate per la frazione in volume:
e analogamente per la molecola B …Nel caso della teoria reticolare delle soluzioni di polimeri, la teoria di Flory-Huggins, valgono le stesse ipotesi che per le soluzioni regolari, fatta eccezione per l’ ipotesi i) in quanto le dimensioni delle macromolecole di polimero sono >> di quelle del solvente. Pertanto, la teoria delle soluz. regolari risulta un caso particolare di quella di Flory-Hugginsper i polimeri, in cui le molec. di soluto e solvente hanno le medesime dimensioni.
BABAAAA uuU φφ += ABABBBB uuU φφ +=
5
Soluzioni di polimeriA differenza delle soluzioni regolari ove le molecole di soluto e solvente hanno uguali dimensioni, - hanno cioè entrambe volume v0, corrispondente al volume di un singolo sito reticolare- le molecole dei polimeri presentano dimensioni >> di quelle di solvente. Nel modello reticolare sviluppato indipendentemente da Flory e Huggins, molecole di pol. e molecole di solv. vengono collocate ancora su di un reticolo, solo che mentre ciasc. mol. di solv. occupa un singolo sito reticolare ciascuna molecola di polimero ne occupa M. In figura M=10.
Supponiamo che il mescolamento continui ad essere casuale, nonostante le interazioni polimero-polimero, solvente-solvente e solvente–polimero possano non essere equivalenti. Inoltre si faccia l’ ipotesi che tutte le catene abbiano la medesima lunghezza M. M rappresenta un rapporto tra due volumi, il volume proprio di ciascuna macromolecola ed il volume di ciasc. molecola di solvente. Esso esprime il num. di di “unità statistiche” di cui è fatta ciascuna molec. di polimero ed è pertanto dello stesso ordine di grandezza del n. di monomeri o grado di polim. della macromolecola. A causa delle grandi differenze di dimensioni delle molecole di ti tipo A e B, piuttosto che adoperare le loro frazioni molari è conveniente adoperare le loro frazioni di volume (φA+φB=1) o di siti occupati, date da:
Il numero di siti totali disponibili è dato data N=MNA+NB, mentre il volume è V= Nv0
BA
AA NMN
MN+
=φBA
BB NMN
N+
=φ
Qual è l’ entropia di mescolamento nell’ ipotesi di mescolamento casuale? Sia ΩΑΒ il numero distinto di modi di collocare NA molecole di polimero e NB molecole di sovente su N siti, ΩΑ il numero di possibili configurazioni distinte relative al sistema A puro, in cui le molecole occupino MNΑ siti e ΩΒ(=1) il numero di configurazioni accessibili al sistema B puro. ∆Sm saràpari a: [ ] [ ]AABBAABm kkS Ω−Ω=Ω−Ω−Ω=∆ lnlnlnlnln
Per ricavare ΩΑΒ immaginiamo di collocare prima le NA molecole di polimero, una per volta e poi le NB molecole di sovente sugli N siti. Supponiamo di aver collocato già i molecolcole di A e di dover collocare la (i+1)-ma. Valutiamo quindi il numero di modi di andare a collocare la (i+1)-ma molecola, ωi+1. Con Mi siti gia riempiti la frazione di siti pieni risulta fi= Mi/N. La prima unità della (i+1)-ma molecola può pertanto essere collocata su N-Mi siti diversi. La prima unità ha z siti primi vicini (z, numero di coordinazione reticolare, pari a 6 nel caso del reticolo cubico semplice). La probabilità che ciascuno di tali siti sia vuota è (1- fi), dal momento che si assume che la distribuzione è casuale (approx. di campo medio). Perciò il num. di possibili ubicazioni per la seconda unità in catena è z(1- fi). La terza unità può andare in (z-1) (1- fi) differenti posti… si ammette che anche le unità 4,5,6…M abbiano a loro volta ciascuna (z-1) (1-fi) possibilità di collocarsi con successo sul reticolo senza andare a collidere con una unità giàcollocata sul reticolo (ciò è un approssimazione,(Flory), ma trattamenti + dettagliati (Huggins) non portano a risultati diversi), per cui, moltiplicando tutti questi fattori avremo per ωi+1:
( ) ( ) ( )1
112
11)1()1(
−
−−−
+−
−=−−−=ω M
MMM
iM
iN
zMiNfzzMiN
Ove si è fatta un ulteriore approx. sostituendo z con (z-1) 6
La quantità ΩΑΒ altri non è che il prodotto (produttoria) di tanti ωi+1 con i=0,2…NA-1 diviso per NA! dal momento che le NA molecole sono indistingibili. Naturalmente, per ciascuna di questa configurazioni in cui tutte le molecole di A sono state già collocate non rimane altro che riempire i siti vuoti con le molecole di B e questo puo’ essere fatto in un unico modo per cui ΩΑΒè dato da:
∏−
=+ω=Ω
1
01!
1 AN
ii
AAB N
Valutiamo ora il logaritmo di detta produttoria…
( ) ( ) ( )∑∏∏−
=
−
=
−−
=+ −+⎟
⎠⎞
⎜⎝⎛ −
−=⎟⎠⎞
⎜⎝⎛ −
−=ω1
0
1
0
11
01 ln1ln11lnln
NMiNM
NzMN
NzMiN
A
iA
AN
i
MM
AN
ii
Dato che NA è molto >>, è possibile considerare i come una variabile continua, per cui siapprossima la sommatoria con un integrale, ottenendo:
( ) ( ) ( )BBB
N
N
NN
iNNNNNN
Mudu
MdiMiNMiN
B
AA
+−−==−≅− ∫∫∑−
=
lnln1ln1lnln0
1
0
( ) ⎟⎠⎞
⎜⎝⎛ −
−+−++−+−=ΩN
zMNNNNNNNNNN ABBBAAAAB1ln1lnlnlnln
Naturalmente nel caso del sistema A puro, N≡M NA per cui lnΩA risulta:
( ) ⎟⎟⎠
⎞⎜⎜⎝
⎛ −−+−++−=Ω
AAAAAAAAA MN
zMNMNMNMNNNN 1ln1lnlnln
[ ] ( )BBAAAABm NNkkS φφ lnlnlnln +−=Ω−Ω=∆ 7
8
( )BBAAm NNkS φφ lnln +−=∆ (1)Tale espressione è identica a quella per la soluzione regolale… ma attenzione, perché mentre nel caso della soluzione regolare la frazione di siti occupata dalle molecole coincide con la frazione molare, nel caso delle soluzioni di polimeri ciò non è + vero ed ha drastiche conseguenze … A titolo di esempio riportiamo in grafico ∆Sm per una soluzione ideale e nel caso di una soluzione di polimero, nell’ ipotesi che M=10 e 500, in funzione di φA. Riscriviamo innanzitutto la (1) come:
⎟⎠⎞
⎜⎝⎛ φφ+φ
φ−=⎟
⎠⎞
⎜⎝⎛ φ+φ−=
∆BBA
AB
BA
AmMN
NN
NkNS
lnlnlnln
Naturalmente, nel caso della soluz. ideale, M=1…L’ entropia di mescolamento di una soluzione regolare in cui molec. di soluto e di solvente abbiano all’ incirca le stesse dimensione si presenta come una curva simmetrica in funzione di φΑ (o φΒ), mentre nel caso delle soluzioni di polimeri tale simmetria si perde.E’ possibile dimostrare che nel caso di una miscela binaria di due polimeri, le cui catene siano costituite da MA e MB unità statistiche, rispettivamente, l’ entropia di mescolamento vale:
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
M=500
MA=MB=10
M=1
∆Sm/k
N
φA
M=10
⎟⎟⎠
⎞⎜⎜⎝
⎛φ
φ+φ
φ−=⎟
⎠⎞
⎜⎝⎛ φ+φ−=
∆B
B
BA
A
AB
BA
AmMMN
NN
NkNS
lnlnlnln
⎟⎟⎠
⎞⎜⎜⎝
⎛+−=⎟
⎠⎞
⎜⎝⎛ +−=
∆=
∆B
B
BA
A
AB
BA
Amm
MMNN
NN
kNS
kS φφφφφφ lnlnlnln
In altri termini l’ espressione:
esprime l’ entropia di mescolamento di una soluzione regolare, e in tal caso MA= MB=1l’ entropia di mescolamento di una soluzione di polimero, e in tal caso MA>>1, MB=1e l’ entropia di mescolamento nel caso di una miscela di due polimeri (blend) nello stato solido, per cui MA>>1, MB>>1.
9
Valutiamo ora l’ energia di mescolamento. Abbiamo già visto che scelto un sito a caso, occupato da una unità di tipo A (oppure B) l’ energia media d’ interazione con l’ unità che occupa uno qualunque dei siti reticolari ad esso primi vicini, UA (UB), risulta l’ energia media d’ interazione tra l’ unità A in questione (oppure B) con tutte le possibilità contemplate dal nostro sistema, cioèil primo vicino può tanto essere una unità di tipo A con probabilità φA (approx. di campo medio), tanto una unità di tipo B con probabilità φA (approx. di campo medio), ovvero:
se l’ unità in questione è di tipo Ase l’ unità in questione è di tipo B.
BABAAAA uuU φφ +=AABBBBB uuU φ+φ=
Dal momento che ciascun sito reticolare occupato da una unità di tipo A ha (z-2) primi vicini (trascurando i terminali che ne hanno (z-1)), l’ energia media di ciascuna unità A con i suoi (z-2) primi vicini è (z-2)UA. Tale è anche l’ energia media di interazione per unità di tipo A con una qualunque unità collocata in un sito primo vicino, e quindi l’ energia media per coppie di siti primi vicini, in cui uno almeno dei siti è occupato da unità di tipo A. Il numero totale di tali coppie sarà NΦA/2 e il contributo all’ energia totale da parte di tali coppie è ½ NΦA(z-2)UA Analogamente, nel caso del solvente, ciascuna unità B ha z primi vicini, per cui l’energia media di ciascuna unità B con i suoi z primi vicini è (z)UB e il contributo all’ energia totale da parte di coppie di siti primi vicini di cui uno almeno sia di tipo B è ½ NφB(z)UB. (Il numero di siti reticolari occupati da unità di tipo A è NΦA, mentre quelli occupati da unità di tipo B sono NφB) . Facciamo una ulteriore approx. ponendo z in luogo di z-2.
10
Sommando tutte le interazioni tra coppie di unità si ottiene l’ energia totale di interazione della miscela e ponendo φ al posto di φΑ e (1-φ) al posto di φB, otteniamo:
[ ] ( ) ( )( ) ( )[ ]
( ) ( )( )[ ]φφφφφ
φφφφφφφφφ
−−+−+=
−+−−+−+=+=
11122
111122
2
2
BBABAA
BABBABAABBAA
uuuzN
uuuuzNUUzNU
Per il componente A puro l’ energia totale di interazione è zuAANAM/2 = zuAANφ/2, mentre per il componente B puro risulta zuBBNB/2 = zuBBN(1-φ)/2, e l’energia totale di entrambe le specie pure è U0=zuAANφ/2+ zuBBN(1-φ)/2. Pertanto l’ energia di mescolamento risulta:
( ) ( )( ) ( )[ ]
( ) ( ) ( )( )[ ]
( ) ( ) ( )[ ]
( )[ ]BBAAAB
BBABAA
BBABAA
BBAABBABAAm
uuuz
uuuz
uuuz
uuuuuzUN
UU
−−−=
−−−+−=
−−−+−+−=
−−−−−+−+=∆=−
212
11212
111122
111122
2
20
φφ
φφφφφφ
φφφφφφ
φφφφφφφ
Si definisce a questo punto il parametro d’ interazione di Flory, χ, per caratterizzare la differenza delle energie di interazione tra le varie specie in soluzione:
( )kT
uuuz BBAAAB −−=
22
χ
11
( )kT
uuuz BBAAAB −−=
22
χ
Il parametro d’ interazione di Flory, χ, è una quantità adimensionale che misura la differenza dell’ energia d’ interazione tra due molecole di specie diverse in una miscela, rispetto all’energia di interazione tra due molecole della stessa specie nei due componenti puri. Usando questa definizione, l’ energia di mescolamento di una soluzione polimerica risulta:
( )kTUm φχφ −=∆ 1Tale equazione esprime l’ energia di campo medio nel caso delle soluzioni binarie regolari, delle soluzioni polimeriche e delle miscele di due polimeri. L’ energia libera di Helmoltz di mescolamento, per unità di siti reticolari risulta pertanto:
( ) ( )⎥⎦
⎤⎢⎣
⎡φ−χφ+φ
φ−+φ
φ−=∆−∆=∆ 1ln1ln B
BA
Ammm MM
kTSTUF
Nel caso delle soluzioni regolari l’ energia libero di Helmoltz di mescolamento vale:
( ) ( )[ ]φχφφφφφ −+−+=∆−∆=∆ 1ln1ln BAmmm kTSTUFNel caso delle soluzioni polimeriche, l’ equazione di Flory Huggins prevede:
( ) ( )⎥⎦⎤
⎢⎣⎡ −+−+=∆−∆=∆ φχφφφφφ 1ln1ln BAmmm M
kTSTUF
Il primo termine è un termine entropico che promuove sempre il mescolamento, anche se nel caso delle miscele polimeriche puo’ essere <<; il secondo termine è un termine energetico e può essere >0, opponendosi alla miscelazione,oppure <0 promuovendo la miscelazione, in dipendenza dal segno di χ
12
Per miscele di specie non polari, che interagiscono fondamentalmente attraverso interazioni dispersive (dipolo indotto-dipolo indotto), il parametro d’ interazione χ di Flory può essere stimato attraverso il metodo sviluppato da Hildebrand e Scott. Esso è basato sul parametro di solubilità δ a sua volta legato all’ energia di vaporizzazione ∆E di una molecola. X es., per la molecola A possiamo scrivere:
con vA il volume della molecola A. Qual è il significato fisico di tale parametro? ∆EA è l’energia di interazione di una molecola A con tutto il suo intorno che è necessario vincere per rimuovere la molecola dallo stato puro Ae isolarla dal suo intorno (vaporizzarla).
A
AA v
E∆=δ
Il rapporto ∆EA/vA rappresenta la densità di energia di coesione ed è l’ energia d’ interazione per unità di volume tra molecole di A puro. L’ energia di interazione per sito nello stato A puro zuAA/2 è legata al parametro di solubilità δA attraverso la relazione:
2002 A
A
AAA vvEvzu
δ=∆
=−
ove v0 è il volume per sito reticolare (il segno meno è introdotto perché l’ energia di vaporizzazione è definita col segno +, mentre l’ energia di interazione uAA è negativa). Analogamente per il componente B avremo:
2002 B
B
BBB vvEvzu
δ=∆
=−
La densità di energia di coesione dovuta a interazioni incrociate A-B è stimata dalla quantità δABstimata attraverso la media geometrica δAB = (δAδB)1/2 per cui avremo:
BAABAB vvzu
δδ=δ=− 02
02
( ) ( )2022
022
2 BABABABBAAAB
kTv
kTv
kTuuuz
δ−δ=δδ−δ+δ
=−−
=χe quindi:
13
Il fatto che χ è in relazione col quadrato della differenza dei parametri di solubilità spiega perchénella maggior parte dei casi χ è >0. Tale approccio funziona bene solo nel caso di interazioni non polari, in cui cioè le interazioni tra omo- ed etero-specie in soluzione sia di tipo Van derWaals e risulta di scarsa validità nel caso di sistemi polari o in presenza di interazioni specifiche, del tipo legami a idrogeno. Le maggiori assunzioni della teoria di Flory Huggins, che: 1- i volumi dei due componenti la miscela o la soluzione sono additivi. 2- le unità di specie A e le unità di specie B hanno lo stesso volume, coincidente con quello del sito reticolare. (Ricordiamo che nel caso delle soluzioni regolari (Hildebrand), le molecole di A e di B sono costituite da una singola unità e MA= MB =1; nel caso delle soluzioni di polimeri (Flory-Huggins) MA>>1, MB =1; nel caso delle miscele polimeriche (Flory) MA>>1, MB >>1).sono ovviamente poco rispecchiate nel caso delle soluzioni reali: Invero, il volume della soluzione (o miscela) cambia all’ atto del mescolamento e si possono verificare effetti locali dipacking… che portano a dover considerare termini indipendenti dalla temperatura additivi nell’espressione del parametro d’ interazione di Flory, quando si confrontano i dati sper. con quelli deducibili dalla teoria. Dal momento che tali effetti non sono in pieno razionalizzabili in termini semplici, quello che si fa, generalmente, è di scaricare tutte le deviazioni dalla teoria nel parametro χ, il quale molto spesso dipende dalla composizione della miscela, dalle lunghezza delle catene polimeriche e dalla temperatura. In pratica χ viene considerato come un parametro EMPIRICO, e si esprime la sua dipend. dalla temperatura attraverso la relazione empirica:
TBA +=χ A è considerato un termine entropico, mentre B/T è la parte entalpica.
14
In pratica la teoria di Flory-Huggins sia per le soluzioni di polimeri, sia per le miscele (o blends) polimeriche) è una teoria largamente usata che funziona molto bene, perché è in grado di prevedere in maniera accurata e precisa lo stato di equilibrio di una miscela, e l’eventuale presenza di una lacuna di miscibilità di un sistema binario al variare della composizione e della temperatura. Cio’ e’sorprendente,date le notevoli approx. e la notevole rozzezza del modello, e il suo valore predittivo si spiega col fatto che in qualche modo riesce a cogliere molto bene gli effetti che governano la termodinamica di equilibrio di tali sistemi, e le approx. fatte per la valutazione dell’ entropia di mesc. vengono alquanto compensate dalle approx. che sottendono alla valutazione del temine energetico. In altri termini, gli errori si compensano, la teoria rimane semplice e soprattutto affidabile. Diamo di seguito i valori di A e B e l’ intervallo di temperatura per alcune coppie di polimeri, spesso adoperate per formare blends, di interesse pratico.
15
16
17
Stabilità ed equilibrio
φ
F
Fαβ
Fα
Fβ
φ0
φα
Fmix
φβ
φ
F
Fαβ
Fα
Fβ
φ0
φα
Fmix
φβ
La stabilità locale di una miscela omogenea di composizione in A Φ0 con energia libera Fmix dipende dalla concavità o convessità locale della curva F vs. Φ. Supponiamo che la soluzione si smescoli in due fasi, l’ una di composizione in A Φα, l’ altra di composizione in A Φβ. L’ ammontare relativo delle due fasi α e β è dato da fα e fβ ove: fα + fβ =1 e Φ0= fαφα + fβφβ = fαφα + (1-fα)φβ = φβ + (φα - φβ) fα
L’ energia libera del sistema a fasi separate Fαβ è dato da (trascurando l’ energia d’ interfaccia tra le due fasi:
Questa dipendenza lineare dell’ energia libera dalla composizione risulta nel segmento di retta evidenziato nelle figure sottostanti AB mentre l’ ammontare relativo delle due fasi è determinato dalla regola della leva, ove si nota che fα= lunghezza del seg. CB/lung. seg. AB mentre fβ = lung. seg. AC/ lung. seg. ΑΒ. La curvatura locale dell’ energia libera determina la stabilità locale della soluzione.
αβ
αβ
αβ
βα φφ
φφφφφφ
−−
=−−
= 00 ; ff
( ) ( )αβ
βααβββαααβ φφ
φφφφ−
−+−=+=
FFFfFfF 00
A
B
B
A
CC
02
2>
Φ∂
∂ F
02
2<
Φ∂
∂ FConcava
Convessa
18
Fmix è maggiore o minore delle di quella del sistema caratterizzato da due fasi separate Fαβ?
φ
F
Fαβ
Fα
Fβ
φ0
φα
Fmix
φβ
φ
F
Fαβ
Fα
Fβ
φ0
φα
Fmix
φβ
A
B
B
A
CC
In questo caso l’ andamento dell’ energia libera vs. φ è concavo, ovvero
per cui Fαβ <Fmix e la soluzione è INSTABILE e tende a smescolarsi. La composizione delle due fasi in equilibrio, èquella per cui il potenziale chimico del componente A nella fase α=potenziale chimico del componente A nella fase β e contemporaneamente il potenziale chimico del componente B nella fase α=potenziale chimico del componente B nella fase β ≡ F MINIMO
02
2
<∂
∂φ
mixF
In questo caso l’ andamento dell’ energia libera vs. φ è convesso, ovvero
per cui Fαβ>Fmix e la soluzione è in equilibrio STABILE.
02
2
>∂
∂φ
mixF
Se la dipend. di F da φ èCONCAVA la soluzione spontaneamente abbassa la propria energia libera separandosi in due fasi…
Se la dipend. di F da φ èCONVESSA la soluz. Èstabile
02
2
<∂
∂φ
mixF INSTABILE
02
2
>∂
∂φ
mixF
Criterio per stabilire la stabilità locale di una miscela:
LOCALMENTE STABILE
19
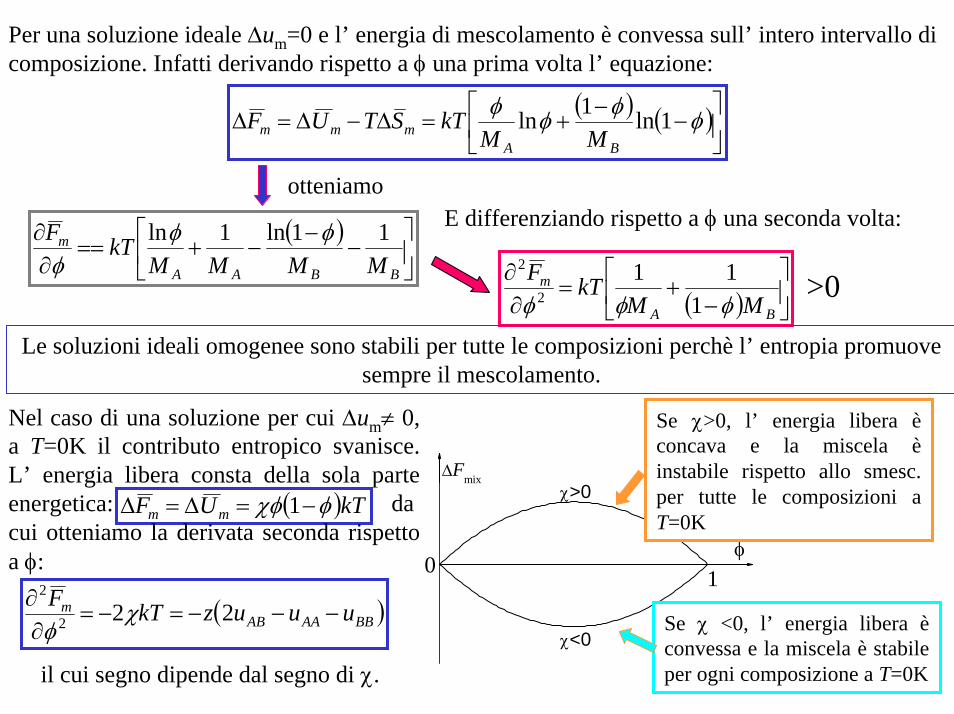
Per una soluzione ideale ∆um=0 e l’ energia di mescolamento è convessa sull’ intero intervallo di composizione. Infatti derivando rispetto a φ una prima volta l’ equazione:
( ) ( )⎥⎦
⎤⎢⎣
⎡−
−+=∆−∆=∆ φφφφ 1ln1ln
BAmmm MM
kTSTUF
otteniamoE differenziando rispetto a φ una seconda volta: ( )
⎥⎦
⎤⎢⎣
⎡−
−−+==
∂∂
BBAA
m
MMMMkTF 11ln1ln φφ
φ
( ) ⎥⎦
⎤⎢⎣
⎡−
+=∂∂
BA
m
MMkTF
φφφ 111
2
2
>0
Le soluzioni ideali omogenee sono stabili per tutte le composizioni perchè l’ entropia promuove sempre il mescolamento.
( )kTUF mm φχφ −=∆=∆ 1
Nel caso di una soluzione per cui ∆um≠ 0, a T=0K il contributo entropico svanisce. L’ energia libera consta della sola parte energetica: da cui otteniamo la derivata seconda rispetto a φ:
( )BBAAABm uuuzkTF
−−−=−=∂∂ 222
2
χφ
il cui segno dipende dal segno di χ.χ<0
φ1
∆Fmixχ>0
Se χ <0, l’ energia libera èconvessa e la miscela è stabile per ogni composizione a T=0K
Se χ>0, l’ energia libera èconcava e la miscela èinstabile rispetto allo smesc. per tutte le composizioni a T=0K
L’ energia libera delle soluzioni reali comporta sia un contributo entalpico sia entropico: la stabilità locale della miscela dipende dal segno della derivata seconda dell’ energia libera rispetto alla composizione:
( ) kTMM
kTF
BA
m χφφφ
21
112
2
−⎥⎦
⎤⎢⎣
⎡−
+=∂∆∂
χΤ=5Κ
φ’ φ”
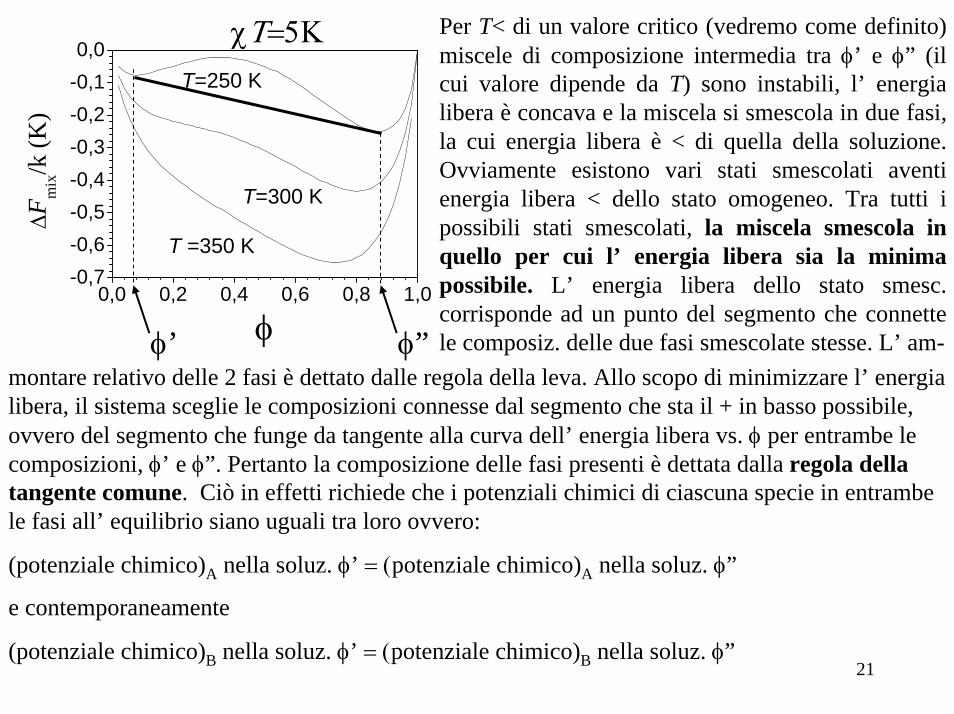
Miscela polimerica asimmetrica con MA=200, MB=100, χ=5/T. Per T sufficientemente >, prevale il termine entropico. Al diminuire di T il termine energetico, che sfavorisce il mescolamento (χ>0), comincia a diventare importante per composizioni della miscela intermedia tra i due estremi. In corrispondenza degli estremi il termine entropico èsempre dominante, per cui la miscela è stabile e non si ha smescolamento. Agli estremi, di fatto, domina sempre il termine entropico rendendo la miscela ricca in A con pochissime molecole di B dissolte o ricca in B con poco A dissolto, stabile, e la curva diventa convessa, ovvero:
Per T< di un valore critico (vedremo come definito) miscele di composizione intermedia tra φ’ e φ” (il cui valore dipende da T) sono instabili, l’ energia libera è concava e la miscela si smescola in due fasi, la cui energia libera è < di quella della soluzione. Ovviamente esistono vari stati smescolati aventi energia libera < dello stato omogeneo. Tra tutti i possibili stati smescolati, la miscela smescola in quello per cui l’ energia libera sia la minima possibile. L’ energia libera dello stato smesc. corrisponde ad un punto del segmento che connette le composiz. delle due fasi smescolate stesse. L’ am-
montare relativo delle 2 fasi è dettato dalle regola della leva. Allo scopo di minimizzare l’ energia libera, il sistema sceglie le composizioni connesse dal segmento che sta il + in basso possibile, ovvero del segmento che funge da tangente alla curva dell’ energia libera vs. φ per entrambe le composizioni, φ’ e φ”. Pertanto la composizione delle fasi presenti è dettata dalla regola della tangente comune. Ciò in effetti richiede che i potenziali chimici di ciascuna specie in entrambe le fasi all’ equilibrio siano uguali tra loro ovvero:
(potenziale chimico)A nella soluz. φ’ = (potenziale chimico)A nella soluz. φ”
e contemporaneamente
(potenziale chimico)B nella soluz. φ’ = (potenziale chimico)B nella soluz. φ”
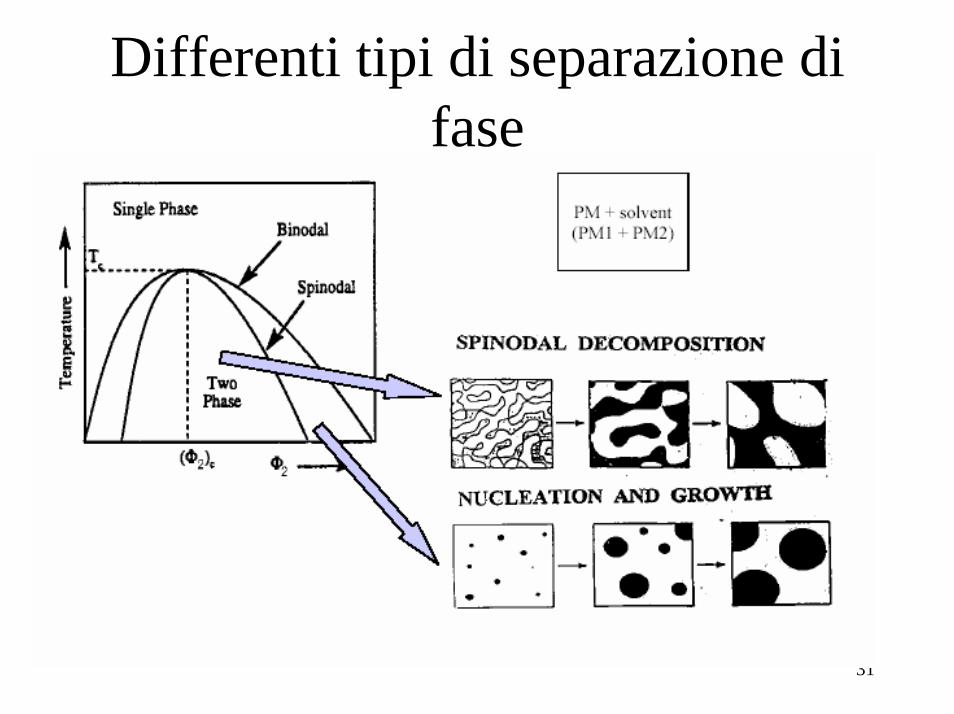
Tra φ’ e φ” e a T=250K, nell’ esempio, la derivata seconda dell’ energia libera, cambia di segno due volte: è > 0 per φ appena maggiori di φ’, poi si annulla per φ=φ(fl1) e diventa <0 per φ>φ(fl1) (la curva dell’ energia libera diventa da convessa concava), quindi si annulla di nuovo per φ=φ(fl2) e quindi ridiventa >0 nuovamente per φ>φ(fl2) (la curva da concava ridiventa convessa). Quindi tra φ’e φ” distinguiamo tre regioni: 1- una regione compresa tra i due flessi per cui la misc. omogenea èinstabile e smescola in due fasi a composizione φ’ e φ” per cui
2- una seconda regione, per composizioni φ’<φ<φ(fl1), in cui: 3- una terza regione, per composizioni φ(fl2)<φ< φ”, in cui:Nelle regione 1 anche piccole fluttuazioni nella composizione della miscela danno luogo ad un abbassamento dell’ energia libera, inducendo lo smescolamento. Tale smescolamento prende il nome di DECOMPOSIZIONE SPINODALE.Nelle regioni 2 e 3, invece, anche se l’ energia libera della miscela è > di quella del sistema a fasi separate, essa risulta stabile rispetto a piccole fluttuazioni della composizione, ma se tali fluttuazioni diventano + grandi, smescola nelle due fasi a composizione φ’ e φ”, attraverso un processo di nucleazione ed accrescimento: solo se si formano “nuclei” delle due fasi di volume abbastanza grande, si innesca lo smescolamento, altrimenti la miscela rimane omogenea, anche se è in uno stato metastabile La decomposizione e’ detta “binodale”.
φ(fl1) φ(fl2)
02
2
>∂∆∂φ
mF
23
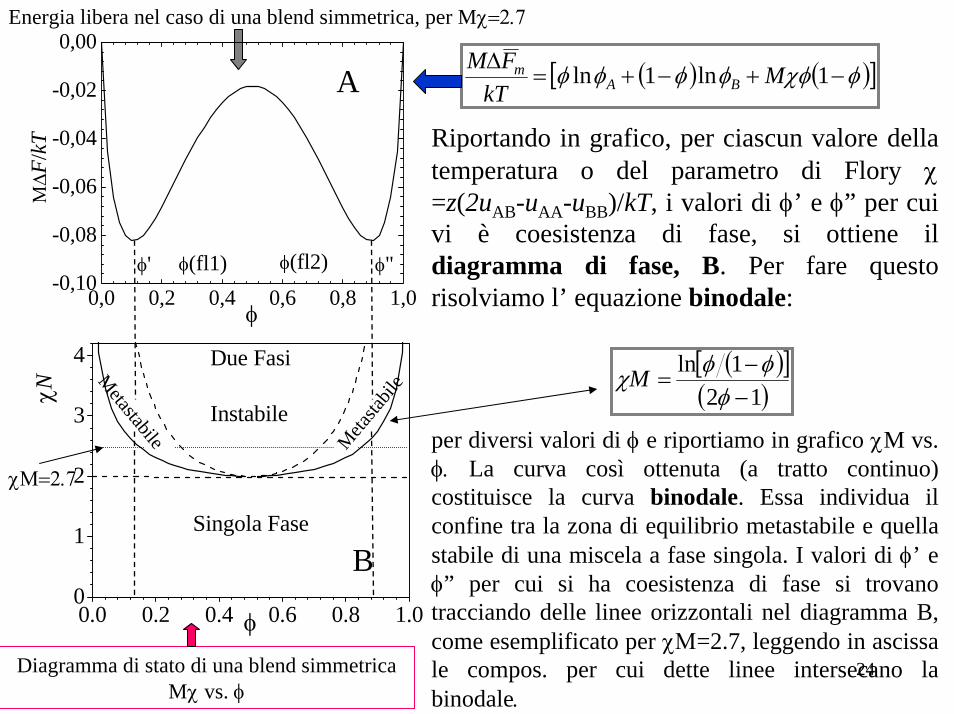
Diagrammi di fase Considerando la dipendenza dell’ energia libera dalla composizione e dalla temperatura èpossibile costruire un diagramma di fase in grado di illustrare il comportamento di fase del dato sistema (nel nostro caso a due componenti), che mostri le regioni (T-composizione) di stabilitàmetastabilità e instabilità. L’ energia libera per un dato sistema binario è data da:
( ) ( )⎥⎦
⎤⎢⎣
⎡−+
−+=∆−∆=∆ φχφφφφφ 1ln1ln B
BA
Ammm MM
kTSTUF
Ad una data temperatura, il sistema esiste in due fasi smescolate piuttosto che sotto forma di una unica fase omogenea, aventi composizioni φ’ e φ” , dettate del requisito di minima energia libera e dalla regola della tangente comune, ovvero:
"' φφφφ φφ ==
⎟⎠
⎞⎜⎝
⎛∂∆∂
=⎟⎠
⎞⎜⎝
⎛∂∆∂ mm FF ( ) ( )⎥
⎦
⎤⎢⎣
⎡φ−χ+−
φ−−+
φ=
φ∂∆∂
2111ln1ln
BBAA
mMMMM
kTFove
Nel caso semplice di una blend simmetrica, MA= MB = M e la tangente comune è un segmento di retta orizzontale (la curva energia libera vs.φ è a sua volta simmetrica) per cui la derivata dell’energia libera rispetto alla composizione del componente A è nulla:
( ) ( ) 0211lnln
"'
=⎥⎦⎤
⎢⎣⎡ −+
−−=⎟
⎠
⎞⎜⎝
⎛∂∆∂
=⎟⎠
⎞⎜⎝
⎛∂∆∂
==
φχφφφφ φφφφ MMFF mm
( )( ) ( )[ ]
( )MMMalebi 121ln1lnln
121
mod −φφ−φ
=⎥⎦⎤
⎢⎣⎡ φ−
−φ
−φ=χ
Ricordando che χ=Α+B/Τ
( )[ ] ( ) AMBT alebi −−φφ−φ
=121lnmod
Equazione binodale Eq. binodale (altra forma)
24
( ) ( )[ ]φχφφφφφ −+−+=∆ 1ln1ln MkT
FMBA
m
Riportando in grafico, per ciascun valore della temperatura o del parametro di Flory χ =z(2uAB-uAA-uBB)/kT, i valori di φ’ e φ” per cui vi è coesistenza di fase, si ottiene ildiagramma di fase, B. Per fare questo risolviamo l’ equazione binodale:
0.0 0.2 0.4 0.6 0.8 1.00
1
2
3
4
Singola Fase
Metastabile
χN
φ
Due Fasi
Instabile
Meta
stabil
e
0,0 0,2 0,4 0,6 0,8 1,0-0,10
-0,08
-0,06
-0,04
-0,02
0,00
φ(fl1) φ(fl2) φ"φ'
Μ
∆F/k
T
φ
χΜ=2.7
A
B
Diagramma di stato di una blend simmetrica Mχ vs. φ
per diversi valori di φ e riportiamo in grafico χM vs. φ. La curva così ottenuta (a tratto continuo) costituisce la curva binodale. Essa individua il confine tra la zona di equilibrio metastabile e quella stabile di una miscela a fase singola. I valori di φ’ e φ” per cui si ha coesistenza di fase si trovano tracciando delle linee orizzontali nel diagramma B, come esemplificato per χΜ=2.7, leggendo in ascissa le compos. per cui dette linee intersecano la binodale.
( )[ ]( )12
1ln−−
=φ
φφχM
Energia libera nel caso di una blend simmetrica, per Mχ=2.7
( ) 02111
2
2=⎥
⎦
⎤⎢⎣
⎡χ−
φ−+
φ=
φ∂
∆∂
BA
mMM
kTF
25( )[ ] ⎥⎦
⎤⎢⎣
⎡−φ−+φ
=AMM
BTBA
spinodale 11121( )⎥⎦
⎤⎢⎣
⎡φ−
+φ
=χ111
21
BAspinadale MM
Ricordando che χ =Α+Β/Τ
Tornando al caso generale in cui MA≠ MB, uguagliando la derivata seconda dell’ energia libera a zero si ottiene l’ equazione della curva spinodale, della curva cioè che delinea il confine tra la regione di metastabilità e quella di instabilità della miscela a fase singola. In altri termini, si trovano le composizioni della miscela per cui i verifica un punto di flesso. Sappiamo che il punto di flesso delinea il confine tra le composizioni della miscela caratterizzate da instabilitàtermodinamica e quelle metastabili.
L’ equazione binodale, che nel caso specifico è:
oppure
consente di trovare per ogni temperatura la coppia di composizioni φ’ e φ” delle due soluzioni in equilibrio in cui smescola spontaneamente una soluzione a composizione intermedia tra questi due valori, rendendo minima l’ energia libera. Ovviamente essa ammette soluzioni solo per alcune temperature, come vedremo, che possono essere > o < di un valore critico, a seconda del valore del parametro χ (o di A e B). Più in generale la curva binodale viene ottenuta risolvendo il sistema di equazioni per cui i potenziali chimici dei due componenti A e B risultano simultaneamente uguali nelle due soluzioni all’ equilibrio. Si dimostra che ciò corrisponde alla situazione di minimo dell’ energia libera. ( ) ( ) ( ) ( ) "'"' ; φφφφ µµµµ BBAA ==
( )[ ] ( ) AMBTbinodale −−φφ−φ
=121ln
( )[ ]( )Mbinodale 12
1ln−φ
φ−φ=χ
26
0.0 0.2 0.4 0.6 0.8 1.00
1
2
3
4
Singola Fase
Metastabile
χM
φ
Due Fasi
Instabile
Meta
stabil
e
0,0 0,2 0,4 0,6 0,8 1,0-0,10
-0,08
-0,06
-0,04
-0,02
0,00
φ(fl1) φ(fl2) φ"φ'
Μ
∆F/k
T
φ
χΜ=2.7
A
B
Diagramma di stato di una blend simmetrica Mχ vs. φ
Riportando in grafico per diversi valori di Mχ (ο Τ) i valori di φ per cui si ha un flesso, si ottiene la curva tratteggiata nel diagramma B, detta curva spinodale, ovvero la curva di confine tra le regioni di instabilità e metastabilità della soluzione omogenea (unica fase).
Energia libera nel caso di una blend simmetrica, per Mχ=2.7
( )⎥⎦⎤
⎢⎣
⎡−
+=φφ
χ1
1121Mspinodale
Punto critico, φχ=0.5, χχΜ=2
In una blend binaria, il punto + basso della curva binodale, coincidente con quello + in basso della curva spinodale, corrisponde al punto critico. Per tale punto si verfica che χ è minimo rispetto a φ:
Che risolta ci dà la concentrazione nel punto critico:
Quest’ ultima relazione sostituita nell’ equazione della spinodale dà la temperatura o il χ critico:
( )0
111
21
22 =⎥⎦
⎤⎢⎣
⎡
−+−=
∂∂
φφφχ
BA MM
27
( ) 2211
21
21
⎟⎟⎠
⎞⎜⎜⎝
⎛+=
⎥⎥⎦
⎤
⎢⎢⎣
⎡ +=
BABA
BAc MMMM
MMχ( ) A
BA
BTBA MMc
c −+=⎥
⎦
⎤⎢⎣
⎡−
= 21121χ
BA
Bc MM
M+
=φ
Nel nostro caso specifico, in cui MA= MB=M, φc=1/2 e χc=2/M
0.0 0.2 0.4 0.6 0.8 1.00
1
2
3
4
Singola Fase
Metastabile
χM
φ
Due Fasi
Instabile
Meta
stabil
e
Diagramma di stato di una blend simmetrica Mχ vs. φ
Punto critico, φχ=0.5, χχΜ=2
Si noti che per valori di χ> χcsussiste una lacuna di miscibilità tra i due rami della curva spinodale; per valori di χ<χc, invece, la miscela risulta omogenea per qualunque composizione.
28
E in una soluzione polimerica?Qui MA=M e MB=1; il diagramma di fase si presenta asimmetrico, con composizione critica:
MMMMM
BA
Bc
11
1≈
+=
+=φ
e cioè risulta molto piccolo, mentre il parametro d’ interazione critico risulta vicino a 1/2:
MMMMM BAc
121121
2111
21
2
+≅⎟⎠
⎞⎜⎝
⎛++=⎟
⎟⎠
⎞⎜⎜⎝
⎛+=χ
Per valori di χ> χc sussiste una lacuna di miscibilità tra i due rami della curva spinodale; per valori di χ<χc, invece, la miscela risulta omogenea per qualunque composizione.
Sperimentalmente, si cambia il parametro d’interazione χ variando la temperatura T. I diagrammi di fase vengono pertanto costruiti in funzione di T e φ. Qui sono mostrati alcuni esempi presi dala letteratura. (A) blendspoli(vinil metil etere)/polistirene(varie masse molec.) (B) Soluzione di poliisoprene in diossano (diverse masse mol.)
29
Se B>0, χ( =Α+Β/Τ) diminuisce all’ aumentare della temperatura. La temperatura + alta della regione bifasica si chiama UCST (upper critical solution temperature), Tc. Per T> Tc la miscela omogenea diventa stabile. Questo è il caso + comune.
Se B<0, χ( =Α+Β/Τ) aumenta all’ aumentare della temperatura. La temperatura + bassa della regione bifasica si chiama LCST (lower critical solution temperature), Tc. Per T< Tc la miscela omogenea diventa stabile. In altri termini qui una miscela omogenea è in grado di dar luogo a separazione di fase all’ aumentare della temperatura.
Vi sono esempi in cui B varia, cambiando di segno, al variare della temperatura… in questo caso il diagramma di fase T-φ è caratterizzato dalle presenza sia di una UCST che di una LCST!
30
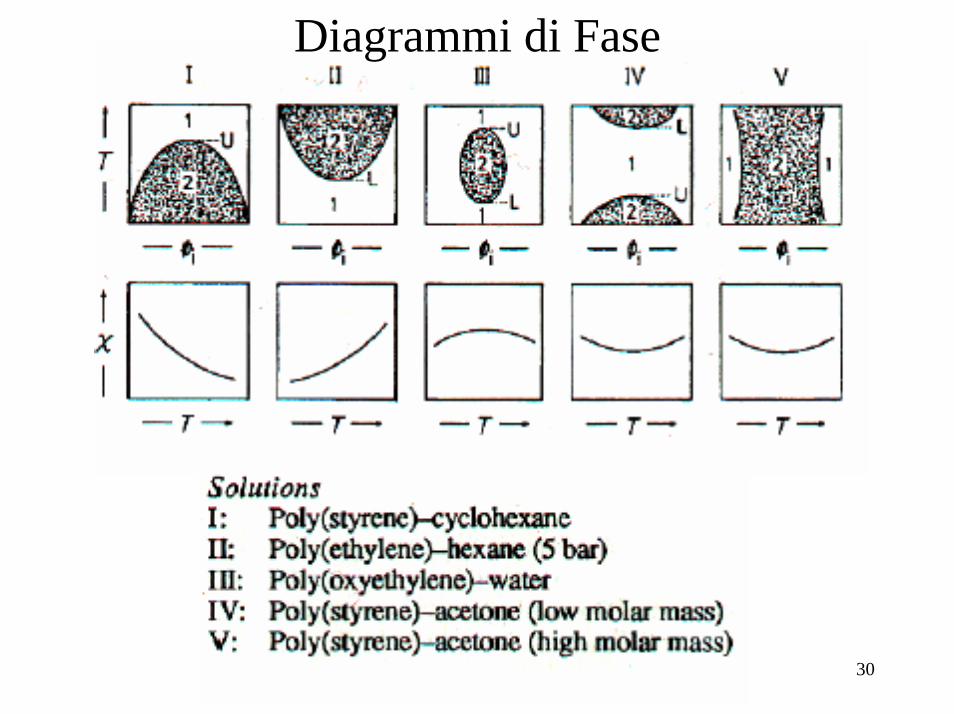
Diagrammi di Fase
31
Differenti tipi di separazione di fase
32
Parametro χ e temperatura θ Per una soluzione polimerica, ove le molecole di polimero siano costituite da M unità statistiche, l’ energia libera molare a volume costante risulta:
⎥⎦⎤
⎢⎣⎡ χφ+φ+φ=∆−∆=∆ BABBA
Ammm NN
MN
kTSTUF lnln
La differenza tra il potenziale chimico del solvente (il componente B la miscela ) in soluzione e quello di B puro risulta la derivata prima dell’ energia libera di Helmoltz rispetto a NB, a temperatura e volume costante:
⎥⎦
⎤⎢⎣
⎡χφ+⎟⎟
⎠
⎞⎜⎜⎝
⎛−φ+φ=µ−µ 20 11ln A
AABBB M
kT
Per soluzioni diluite φΑ << 1, per cui è possibile espandere in serie il termine ln φΒ = ln(1-φΑ)=−φΑ− φΑ
2/2− φΑ3/3-… ottenendo:
⎥⎦
⎤⎢⎣
⎡φ⎟
⎠⎞
⎜⎝⎛ −χ+
φ−=µ−µ 20
21
AA
ABB M
kT
Il termine φA/MA esprime l’ effetto sul potenziale chimico del solvente puro, µB0, della presenza
di un soluto (ovvero il pot. chimico del solvente aumenta di tale quantità perche’ c’e’ il soluto). Il termine in φA
2 dipende dalle interazioni tra coppie di segmento polimero-polimero (effetto volume escluso). Poiché per un cattivo solvente χ>0 per annullare l’ effetto VOLUME ESCULSO è necessario che χ=1/2 e che quindi sia leggermente cattivo. Tale è il solvente THETA (θ).
33
Più in generale, ricordando che χ =A+B/T, alla temperatura θ otteniamo che 1/2=A+B/θ da cui si ricava che B/θ = 1/2 -A e quindi
θ = B/ (1/2 -A )=B/ ψ ove ψ=(1/2−Α). Riscriviamo ora χ −1/2 sotto forma di (χ −1/2) =A+B/T-1/2
sostituendo: A=1/2-ψ e B= θψ avremo:
(χ −1/2) =A + B/T - 1/2=1/2 - ψ + θψ/T - 1/2 = ψ(θ − T) /TGeneralmente Α è <1/2 (in valore assoluto) e B>0, per cui ψ>0. Se allora T> θ, (χ −1/2)<0, per cui la differenza tra il potenziale chimico del solvente nella soluzione e quello del solvente puro diminuisce a tali temperature:
Il solvente diventa un buon solvente, in questo caso, a T>θ.Per T< θ invece, (χ −1/2)>0, e il solvente a tali temperature può diventare cattivo. La temperatura a cui diventa cattivo è tanto + vicina a quella theta quanto maggiore è MA. Per solventi polari che sciolgono a loro volta soluti polari può succedere che ψ<0, pur rimanendo la temperatura theta definita in quanto anche B<0 (temperature theta <0 non hanno significato fisico, perche’ minori dello zero assoluto). In questo caso il solvente èbuono per T< θ,(( χ −1/2)<0) e può diventare cattivo per T> θ,( (χ −1/2)>0).
⎥⎦
⎤⎢⎣
⎡φ⎟
⎠⎞
⎜⎝⎛ −χ+
φ−=µ−µ 20
21
AA
ABB M
kT
34
Effetto volume escluso,condizioni theta e fusi polimerici-Ipotesi di Flory
Abbiamo precedentemente ricavato che le dimensioni di una macromolecola in un solvente thetacorrispondono a quelle di una catena ideale che non risente affatto del VOLUME escluso. In questo caso Rg, la radice quadrato del raggio di girazione quadratico medio rispetta la legge di proporzionalità:
con l la lunghezza di legame e n il numero di legami che costituiscono la catena e si dice che la catena è nello stato imperturbato. Ciò è una diretta conseguenza del fatto che i modelli che forniscono tale legge di proporzionalità prendono in considerazione al + interzioni di volume escluso a corto raggio (modello RIS) ma trascurano le interazioni di volume escluso a lungo raggio. In altri termini porzioni di catena distanti tra loro parecchi legami (+ di 4) ignorano la loro reciproca presenza nello spazio per cui possono occupare la stessa posizione.
2/1nlRg ∝
La catena isolata nel vuoto non potrebbe mai e poi mai, se potesse esistere, rispondere a detta legge di proporzionalità. Infatti, ragionando per assurdo, la conformazione di detta catena dovrebbe in questa realtà virtuale essere piu’ espansa di quella prevedibile in base a tale legge. Immaginiamo di creare noi tale catena aggiungendo una unità monomerica per volta a partire da un certo punto nello spazio… succede che ad ogni step di crescita, ci saranno mediamente piùunità monomeriche dietro all’ ultima unità aggiunta che davanti a questa. In altri termini, la catena guadagna + entropia accrescendosi in avanti piuttosto che accrescendosi tornando indietro, per cui viene ad assumere, in maniera naturale, dimensioni maggiori di quelle dell’ ipotetica catena che non risente affatto durante la “crescita” della presenza dei suoi predecessori.
35
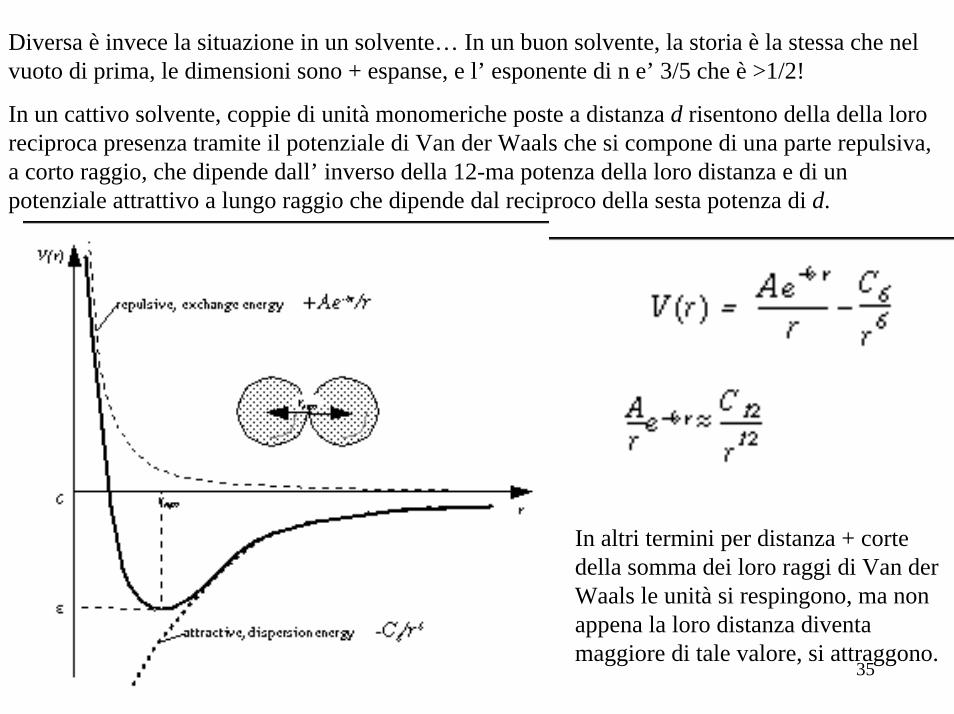
Diversa è invece la situazione in un solvente… In un buon solvente, la storia è la stessa che nel vuoto di prima, le dimensioni sono + espanse, e l’ esponente di n e’ 3/5 che è >1/2!
In un cattivo solvente, coppie di unità monomeriche poste a distanza d risentono della della loro reciproca presenza tramite il potenziale di Van der Waals che si compone di una parte repulsiva, a corto raggio, che dipende dall’ inverso della 12-ma potenza della loro distanza e di un potenziale attrattivo a lungo raggio che dipende dal reciproco della sesta potenza di d.
In altri termini per distanza + corte della somma dei loro raggi di Van derWaals le unità si respingono, ma non appena la loro distanza diventa maggiore di tale valore, si attraggono.
36
Ipotizziamo sempre che la macromolecola si accresca aggiungendo una unità per volta, gradualmente. Poiché si accresce in un ambiente ostile, la sua direzione di crescità saràpreferenzialmente nella direzione delle unità già presenti, piuttosto che dal fronte opposto per cui due unità possono venirsi a trovare a distanza tali da attrasi in maniera significativa. Se questa attrazione è forte abbastanza perché il solvente non ne vuole sapere di “avere a che far”e con la macromolecola, le dimensioni della stessa diventano < di quelle della catena nello stato imperturbato e scalano come n1/3. Naturalmente tra il cattivo solvente e il buon solvente c’è anche quello che essendo “cattivello” che fa si’ che mediamente unità separate da molti legami stiano piu’ vicine tra loro rispetto alla catena che si accresce nel vuoto, in maniera che si attraggano quel tanto da annullare del tutto le interazioni repulsive a corto raggio… La molecola assume le dimensioni dello stato imperturbato in questo solvente e il solvente e quello theta. Dalla teoria abbiamo visto infatti che questo e’ effettivamente cattivello, in quanto χ=1/2.
Veniamo ora all’ ipotesi di Flory, che ha acquistato notevole credibilità perche’ e’ stata largamente confermata dai fatti… che cosa succede in un fuso polimerico? Qui ciascuna unità è circondata isotropicamente da altre unità e non ha modo di decidere se quelle che la circondano appartengano alla stessa catena oppure a catene diverse…. ERGO non ci sarà alcuna direzione preferenziale di crescita della catena stessa (nell’ ipotetica operazione che questa si crei nell’ ambiente aggiungendo un monomero per volta) e la macromolecola assume le dimensioni dello stato imperturbato o theta.