77
1 VALUTAZIONE DEL DATO ANALITICO VALUTAZIONE DEL DATO ANALITICO Università Ca’ Foscari di Venezia Dipartimento di Chimica Fisica Dr. Ligia Maria Moretto AA. 2006/07

1
VALUTAZIONE DEL DATO ANALITICO VALUTAZIONE DEL DATO ANALITICO
Università Ca’ Foscari di Venezia
Dipartimento di Chimica Fisica
Dr. Ligia Maria MorettoAA. 2006/07
2
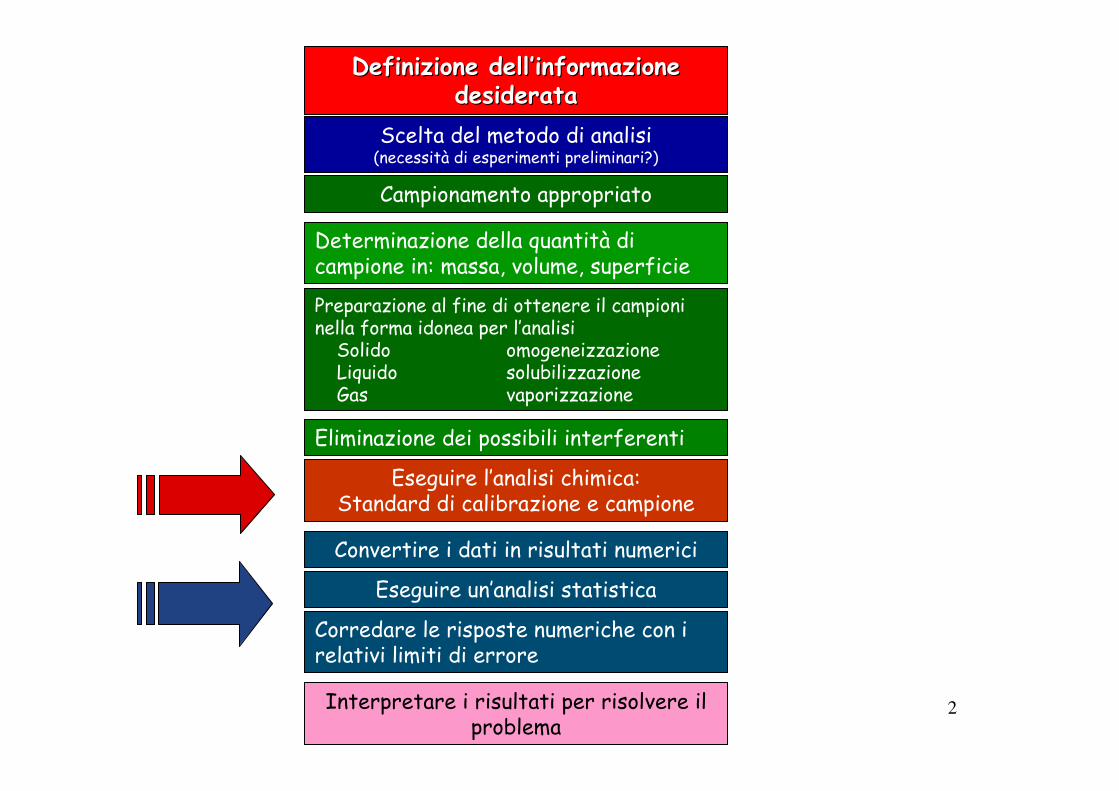
Definizione dellDefinizione dell’’informazione informazione desideratadesiderata
Scelta del metodo di analisi(necessità di esperimenti preliminari?)
Campionamento appropriato
Determinazione della quantità di campione in: massa, volume, superficie
Preparazione al fine di ottenere il campioni nella forma idonea per l’analisi
Solido omogeneizzazioneLiquido solubilizzazioneGas vaporizzazione
Eliminazione dei possibili interferenti
Eseguire l’analisi chimica: Standard di calibrazione e campione
Convertire i dati in risultati numerici
Interpretare i risultati per risolvere il problema
Eseguire un’analisi statistica
Corredare le risposte numeriche con i relativi limiti di errore

3
L’importanza della valutazione del dato analitico ottenuto in
laboratorio
LL’’importanza della valutazione importanza della valutazione del dato analitico ottenuto in del dato analitico ottenuto in
laboratoriolaboratorio
4
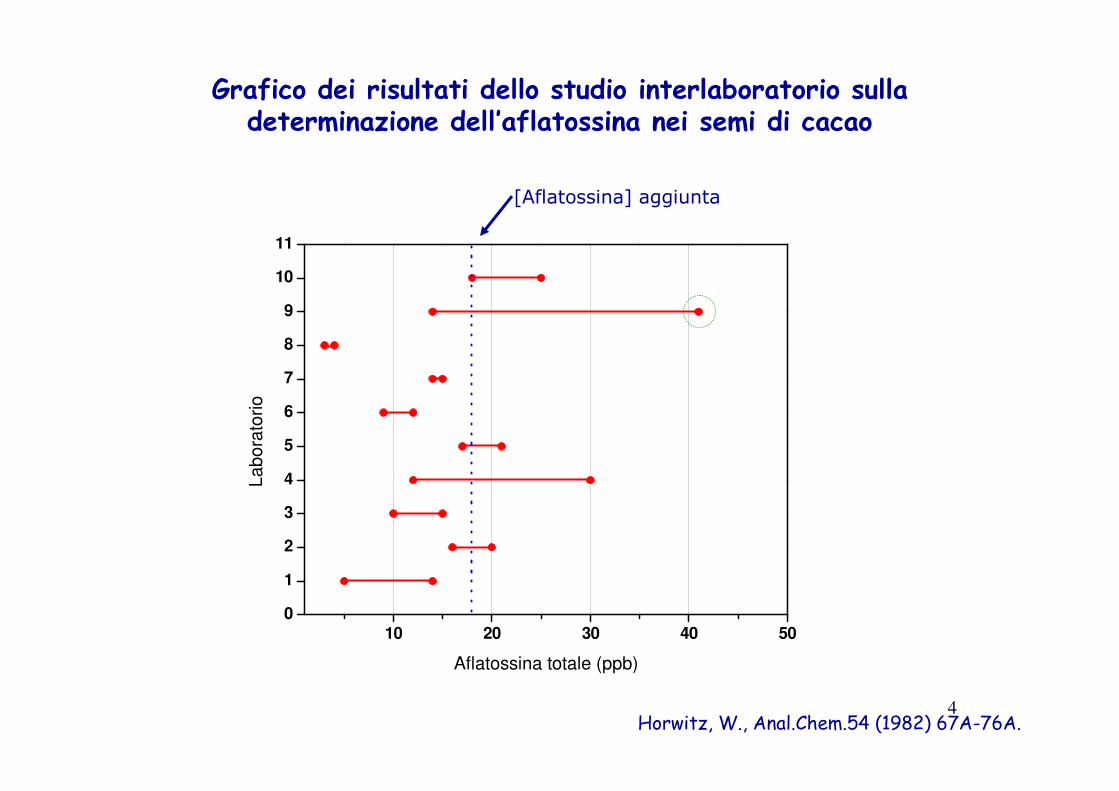
Grafico dei risultati dello studio interlaboratorio sulla determinazione dell’aflatossina nei semi di cacao
Horwitz, W., Anal.Chem.54 (1982) 67A-76A.
0
1
2
3
4
5
6
7
8
9
10
11
10 20 30 40 50
Aflatossina totale (ppb)
La
bo
rato
rio
[Aflatossina] aggiunta

5
Valutazione dei dati analiticiValutazione dei dati analitici
Replicati: campioni identici analizzati esattamente nello stesso modo.
Media: la media di due o più misure si identifica con la media aritmetica.
N
x
x
N
i
i∑== 1
Mediana: la mediana è il valore centrale di un set di dati che sono stati ordinati in ordine di grandezza. E’ usata spesso quando un set contiene un outlier, cioè un valore che differisce significativamente del resto dei dati. Un outlier può avere un effetto marcato sulla media, ma non sulla mediana..

6
ACCURATEZZA: grado di concordanza tra valore misurato e valore vero.
Viene espresso il termini di ERRORE - normalmente errore percentuale.
Valore “vero” di una misura: in generale è sconosciuto.
Per determinare l’accuratezza di un metodo analitico si può ricorrere a:
-materiali di riferimento certificati
-confronto con altri metodi d’analisi standard
- intercalibrazione tra laboratori diversi

7
PRECISIONE: grado di concordanza tra misurazioni diverse di un campione eseguite nello stesso modo.
Si determina mediante analisi replicata.E’ espressa mediante la DEVIAZIONE STANDARD.

8
RIPETIBILITA’: analisi replicata del campione nelle stesse identiche condizioni all’interno di un set di misure (within-run precision)
RIPRODUCIBILITA’: analisi replicata del campione in condizioni diverse (es. diverso giorno, diverso operatore, diverso reagente, etc) (between-run precision)
RipetibilitRipetibilitàà e riproducibilite riproducibilitàà sono due tipi di precisione: sono due tipi di precisione:
9
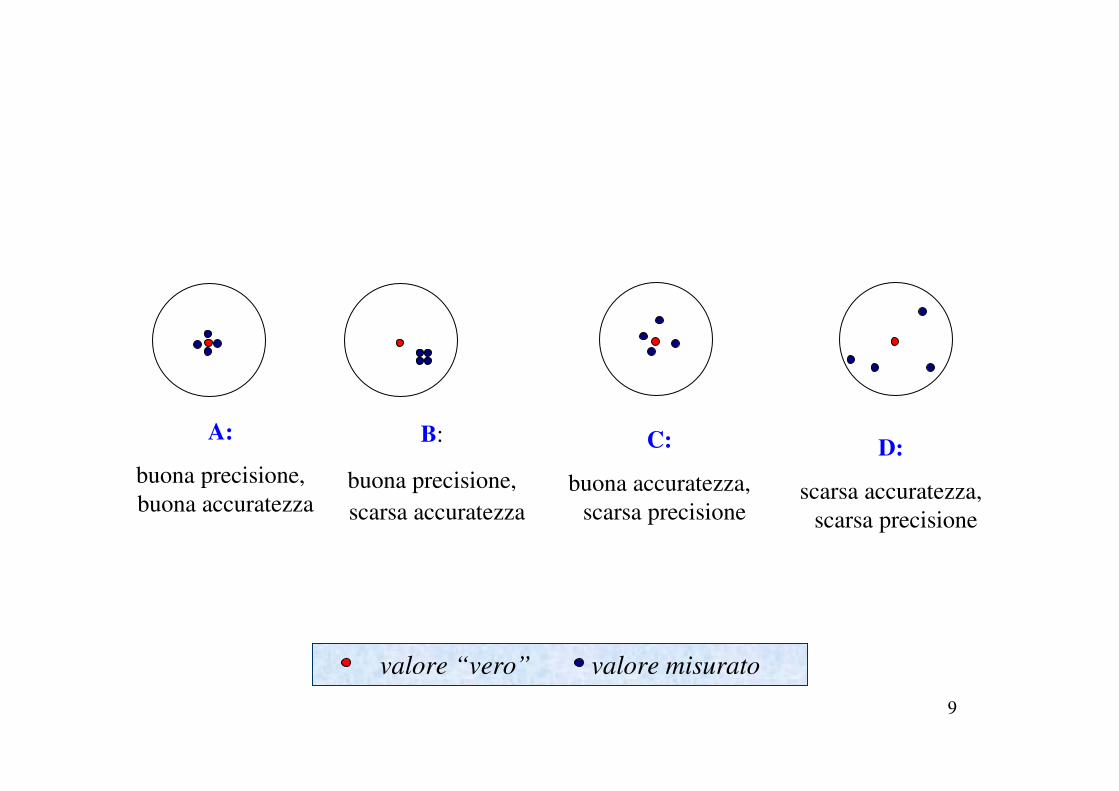
A:
buona precisione,
buona accuratezza
B:
buona precisione,
scarsa accuratezza
C:
buona accuratezza,
scarsa precisione
D:
scarsa accuratezza,
scarsa precisione
valore “vero” valore misurato

10
Cifre significative
Dato analitico Da misura sperimentale
Risultato di una prova Numero ottenuto dopo elaborazioni, calcoli, ecc
Cifre significativePrestazioni degli strumenti (incertezza della misura)
Dal metodo usato
Riportare le cifre significative note con la certezza piùla prima cifra incerta indicando l’intervallo di
incertezza.
Riportare le cifre significative note con la certezza piùla prima cifra incerta indicando l’intervallo di
incertezza.

11
Esempio di grandezze e relative incertezze:Esempio di grandezze e relative incertezze:
Bilancia digitale con precisione di ± 0.1 mg 4.0057 ± 0.0001 g
Bilancia digitale con precisione di ± 0.02 g 4.00 ± 0.02 g
Potenziometro digitale con precisione di ± 1mV 434 ± 1 mV
Potenziometro analogico con precisione di ± 5mV 434 ± 5 mV
Buretta con divisioni da 0.05 mL e tolleranza di ± 0.03 mL 5.25 ± 0.03 mL
Spettrofotometro con precisione di ± 0.001 A 0.897 ± 0.001 A
Massa molare di Na2C2O4 134.01 ± 0.1

12
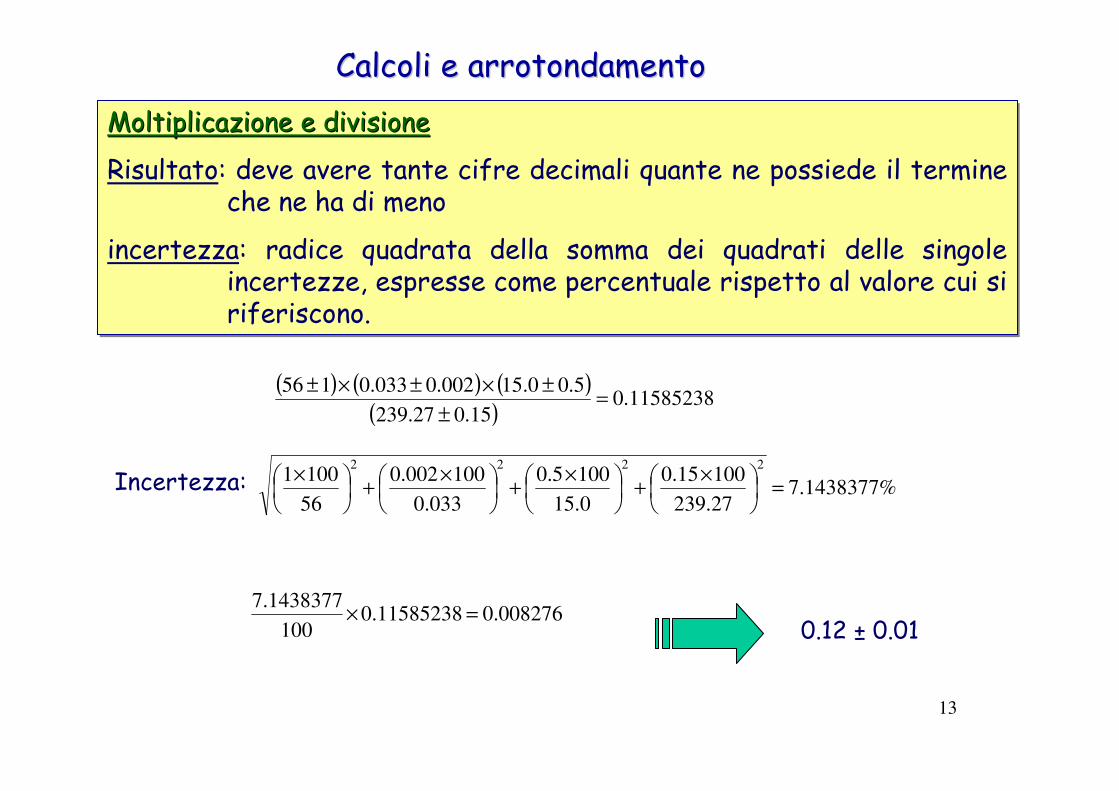
Calcoli e arrotondamentoCalcoli e arrotondamento
Addizione e sottrazioneAddizione e sottrazione
Risultato: deve avere tante cifre decimali quante ne possiede il termine che ne ha di meno
incertezza: uguale alla radice quadrata della somma dei quadrati delle singole incertezze
(10.1 ± 0.2)+ (223.23 ± 0.01)+ (1456.72 ± 0.05) = 1690.05
1690.0 ± 0.2
( ) ( ) ( ) 2026.005.001.02.0222
=++Incertezza:
13
Calcoli e arrotondamentoCalcoli e arrotondamento
Moltiplicazione e divisione
Risultato: deve avere tante cifre decimali quante ne possiede il termine che ne ha di meno
incertezza: radice quadrata della somma dei quadrati delle singole incertezze, espresse come percentuale rispetto al valore cui si riferiscono.
Moltiplicazione e divisioneMoltiplicazione e divisione
Risultato: deve avere tante cifre decimali quante ne possiede il termine che ne ha di meno
incertezza: radice quadrata della somma dei quadrati delle singole incertezze, espresse come percentuale rispetto al valore cui si riferiscono.
0.12 ± 0.01
( ) ( ) ( )( )
11585238.015.027.239
5.00.15002.0033.0156=
±
±×±×±
Incertezza: %1438377.727.239
10015.0
0.15
1005.0
033.0
100002.0
56
10012222
=
×+
×+
×+
×
008276.011585238.0100
1438377.7=×

14
Errore assoluto di una misura è la differenza, compreso il segno, tra il valore misurato (xi) e il valore vero (xt)
ti xxE −=
Errore relativo di una misura è dato dall’errore assoluto diviso per il valore vero
%100×−
=t
ti
rx
xxE
Tipi di errori nei dati sperimentali
1. errori sistematici (o determinati): influenzano l'accuratezza
2. errori casuali (o indeterminati): influenzano la precisione
3. errori grossolani (responsabili per gli outliers)
Tipi di errori nei dati sperimentaliTipi di errori nei dati sperimentali
1. errori sistematici (o determinati): influenzano l'accuratezza
2. errori casuali (o indeterminati): influenzano la precisione
3. errori grossolani (responsabili per gli outliers)

15
1. Errori sistematici:1. Errori sistematici:Errori strumentali: causati da malfunzionamento degli strumenti, strumenti starati, instabilità sorgenti di energia, ecc.
controllo: calibrazione periodica degli strumenti, ecc
Errori di metodo: sono i più difficili da identificare e correggere. Es: incompletezza di reazioni, perdita per evaporazione, adsorbimento di analita, interferenti, ecc.
Rivelazione: analisi di campione standard, analisi indipendenti, determinazione del bianco, usare metodo analitico diverso.
Errori personali: es: stima posizione indicatore, colore di una soluzione, livello del menisco di un liquido, ecc.
Minimizzazione:attenzione e autodisciplina, controllo dati di partenza, calcoli, ecc.

16
2. Errori casuali:2. Errori casuali:
Errore casuale o errore indeterminato: Deriva dall’effetto prodotto dalla presenza di variabili incontrollate (o incontrollabili) nelle misure.L’errore casuale ha identiche probabilità di essere positivo o negativo.
La statistica fornisce strumenti utili per decidere di accettareconclusioni che hanno elevata probabilità di essere corrette e rifiutare altre che non ne hanno.
La statistica fornisce strumenti utili per decidere di accettareconclusioni che hanno elevata probabilità di essere corrette e rifiutare altre che non ne hanno.
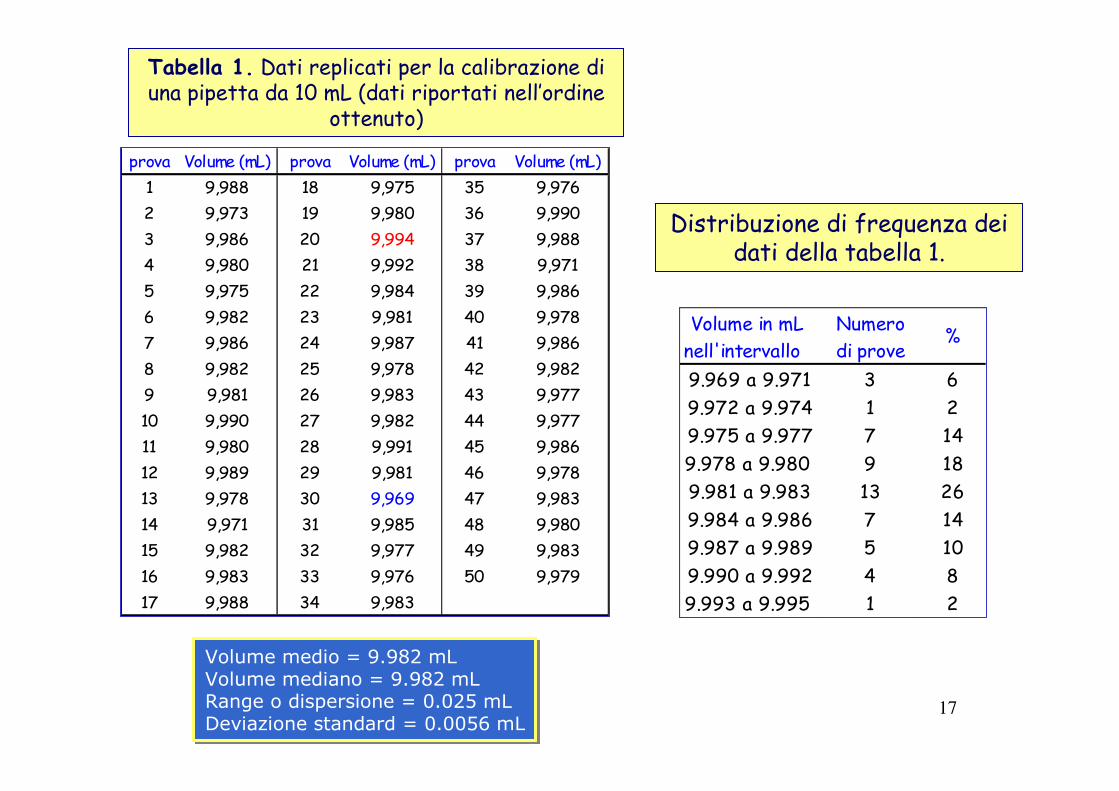
17
prova Volume (mL) prova Volume (mL) prova Volume (mL)1 9,988 18 9,975 35 9,9762 9,973 19 9,980 36 9,9903 9,986 20 9,994 37 9,9884 9,980 21 9,992 38 9,9715 9,975 22 9,984 39 9,9866 9,982 23 9,981 40 9,9787 9,986 24 9,987 41 9,9868 9,982 25 9,978 42 9,9829 9,981 26 9,983 43 9,97710 9,990 27 9,982 44 9,97711 9,980 28 9,991 45 9,98612 9,989 29 9,981 46 9,97813 9,978 30 9,969 47 9,98314 9,971 31 9,985 48 9,98015 9,982 32 9,977 49 9,98316 9,983 33 9,976 50 9,97917 9,988 34 9,983
Volume medio = 9.982 mLVolume mediano = 9.982 mLRange o dispersione = 0.025 mLDeviazione standard = 0.0056 mL
Volume medio = 9.982 mLVolume mediano = 9.982 mLRange o dispersione = 0.025 mLDeviazione standard = 0.0056 mL
Volume in mL Numeronell'intervallo di prove9.969 a 9.971 3 69.972 a 9.974 1 29.975 a 9.977 7 149.978 a 9.980 9 189.981 a 9.983 13 269.984 a 9.986 7 149.987 a 9.989 5 109.990 a 9.992 4 89.993 a 9.995 1 2
%
Distribuzione di frequenza dei dati della tabella 1.
Tabella 1. Dati replicati per la calibrazione di una pipetta da 10 mL (dati riportati nell’ordine
ottenuto)
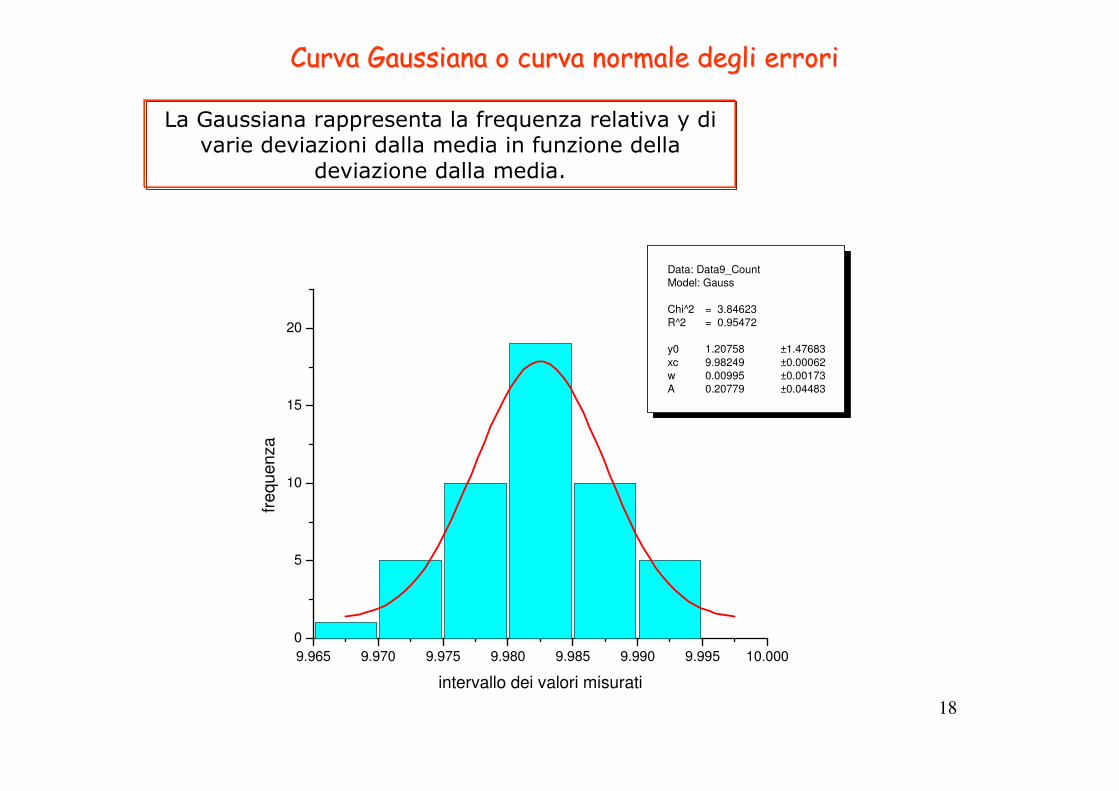
18
Curva Curva GaussianaGaussiana o curva normale degli errorio curva normale degli errori
La Gaussiana rappresenta la frequenza relativa y di varie deviazioni dalla media in funzione della
deviazione dalla media.
9.965 9.970 9.975 9.980 9.985 9.990 9.995 10.000
0
5
10
15
20
Data: Data9_Count
Model: Gauss
Chi^2 = 3.84623
R^2 = 0.95472
y0 1.20758 ±1.47683
xc 9.98249 ±0.00062
w 0.00995 ±0.00173
A 0.20779 ±0.04483
frequenza
intervallo dei valori misurati
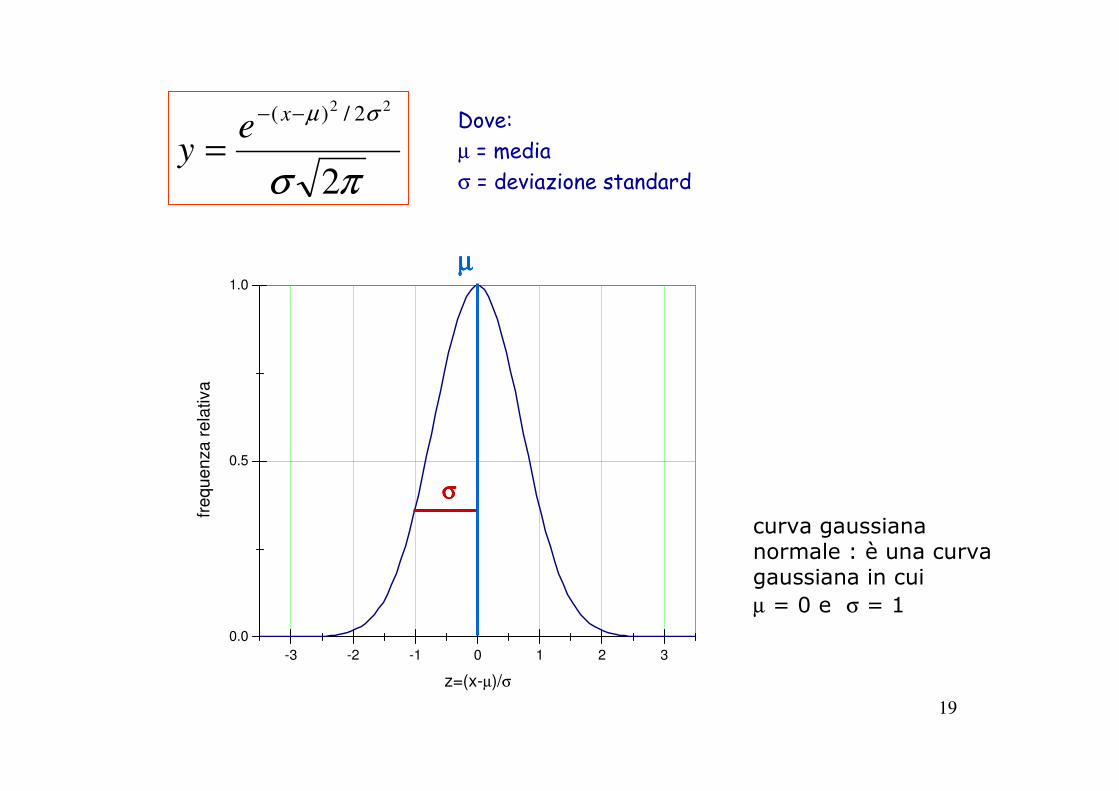
19
curva gaussiananormale : è una curva gaussiana in cui
µ = 0 e σ = 1
-3 -2 -1 0 1 2 3
0.0
0.5
1.0
fre
qu
en
za
re
lativa
z=(x-µ)/σ
σσσσ
µµµµ
πσ
σµ
2
22 2/)( −−
=x
ey
Dove:µ = mediaσ = deviazione standard
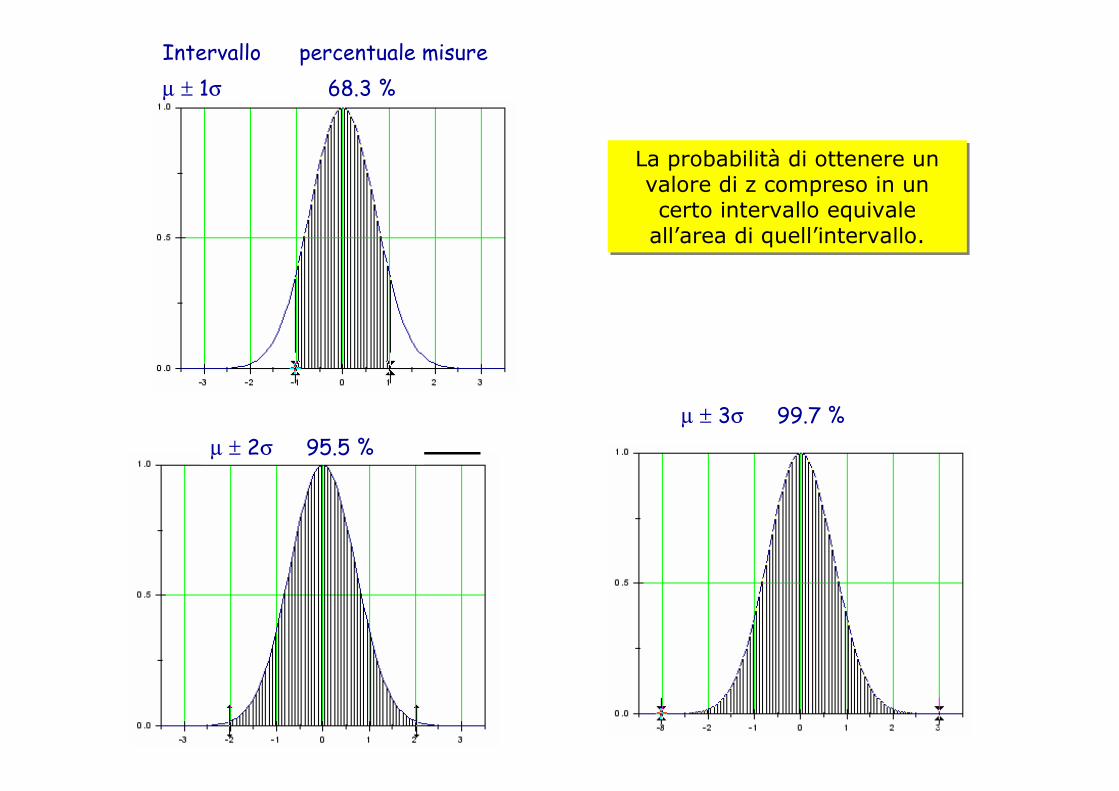
20
La probabilità di ottenere un valore di z compreso in un certo intervallo equivale all’area di quell’intervallo.
La probabilità di ottenere un valore di z compreso in un certo intervallo equivale all’area di quell’intervallo.
Intervallo percentuale misureµ ± 1σ 68.3 %
µ ± 2σ 95.5 %µ ± 3σ 99.7 %
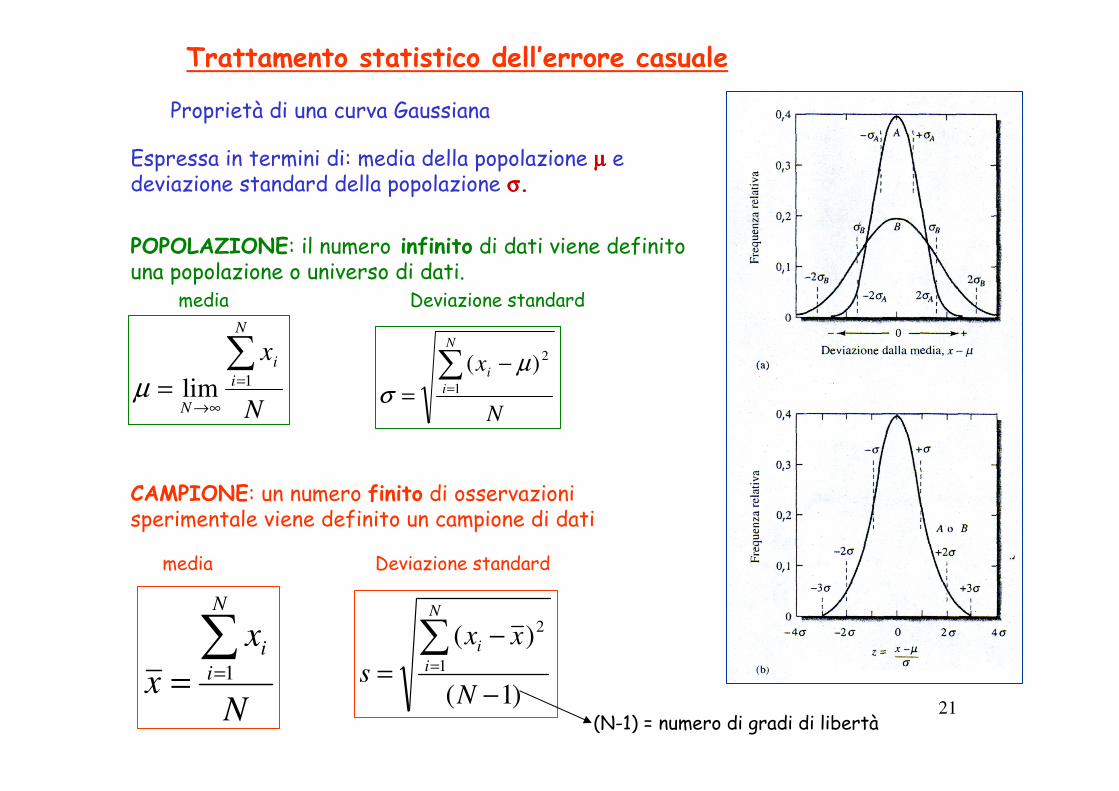
21
Trattamento statistico dell’errore casuale
Espressa in termini di: media della popolazione µµµµ e deviazione standard della popolazione σσσσ.
Proprietà di una curva Gaussiana
POPOLAZIONE: il numero infinito di dati viene definito una popolazione o universo di dati.
N
xN
i
i
N
∑=
∞→= 1limµ
N
xN
i
i∑=
−
= 1
2)( µ
σ
media Deviazione standard
CAMPIONE: un numero finito di osservazioni sperimentale viene definito un campione di dati
N
x
x
N
i
i∑== 1
media Deviazione standard
)1(
)(1
2
−
−
=∑
=
N
xx
s
N
i
i
(N-1) = numero di gradi di libertà

22
Misura della precisioneMisura della precisione
)1(
)(1
2
−
−
=
∑=
N
xx
s
N
i
iDeviazione standard del campione:
Varianza:1
)( 2
12
−
−
=
∑=
N
xx
s
N
i
i
Deviazione standard relativa: 1000)(x
sRSD =
Coefficiente di variazione: 100)(x
sCV =

23
Esempio:
Nel corso di una analisi replicata relativa al contenuto di piombo nel sangue, sono stati ottenuti i seguenti risultati:
0.752 0,756 0,752 0.751 0.760 ppm di Pb
Calcolare la media e la deviazione standard di questo insieme di dati.
Eq. 3.4 : riarrangiamentodell’equazione di s
1
)(
1
1
2
2
−
−
=
∑∑
=
=
N
N
x
x
s
N
i
N
i
i
i
Fare con foglio excell o origin!Fare con foglio excell o origin!

24
Per l’insieme dei dati dell’esempio precedente, calcolare:a) la varianzab) la deviazione standard relativa in parti per millec) il coefficiente di variazioned) la dispersione
X = 0.754 ppm Pb s = 0.0038 ppm Pb
A) s2 = (0.0038)2 = 1.4x10-5
B) RSD = (0.0038/0.754)x1000 = 5.0 ppt
c) CV = (0.0038/0.754)x100 = 0.5%
D) w = 0.760 - 0.751 = 0.009
0,0000142varianza
0,0037683sdt dev
0,7542media
0,752
0,760
0,751
0,756
0,752

25
Si esegue un’analisi per la determinazione delle proteine in un lotto di proteina nitrogenasi ferro-molibdeno in soluzione 0.1 M NaCl pH 7.35 con tampone tris. I valori trovati per i cinque campioni sono: 27.5, 28.3, 29.0, 28.5 e 28.2 mg di proteina per mL. Calcolare il valore medio della concentrazione della proteina e la mediana delle misure, la deviazione standard, la deviazione standard relativa e la dispersione.
Mediana: 28.3 mg/L Dati ordinati: 27.5, 28.2, 28.3, 28.5, 29.0
N
x
x i
i∑= 28.3mg/L
5
28.2)28.529.028.3(27.5x =
++++=Media:
Deviazione standard:
)1(
)(1
2
−
−
=
∑=
N
xx
s
N
i
i0.54 mg/L
Deviazione standard relativa: RSD = (s/x)100 = (28.3/0.54)x100= 1.9%
Dispersione: w = 29.0-27.5 = 1.5 mg/L

26
LL’’affidabilitaffidabilitàà di s come misura della precisionedi s come misura della precisione
Raccolta di dati per aumentare l’affidabilità di s
Deviazione standard cumulata
t
N
i
N
j
ji
cumulataNNN
xxxx
s−++
+−+−
=
∑ ∑= =
...
...)()(
21
1 1
2
2
2
1
1 2
Dove: N1 è il numero di dati nell’insieme 1,N2 è il numero di dati nell’insieme 2, ecc. Nt è il numero degli insiemi di dati che sono campionati.

27
E’ stato determinato il mercurio in sette pesci pescati nella Baia di Chesapeakecon un metodo basato sull’assorbimento di una radiazione da parte del mercurio gassoso elementare. Calcolare una stima accumulata della deviazione standard per il metodo utilizzato, basata sulle prime tre colonne di dati:
0.2196Somma dei
quadrati
28N°misure
0.01701.1301.11, 1.15, 1.22, 1.04
47
0.06852.4822.35, 2.44, 2.70,
2.48,2.44
56
0.01140.5700.57,0.58, 0.64, 0.49
45
0.06112.0182.06, 1.93, 2.12, 2.16, 1.89,1.95
640.02423.2403.13, 3.3523
0.01151.0150.96, 0.98, 1.02, 1.10
42
0.02581.6731.80,1.58,1.64
31
Somma quadrati
deviazione dalla media
[Hg]Media
(ppm)
[Hg]
(ppm)
N°campioni analizzati
Campione
Calcolo della media (4° colonna):
X= (1.80+1.58+1.64)/3= 1.673
Somma dei quadrati della deviazione della media:
(1.80-1.673)2+(1.80-1.,58)2+(1.80-1.64)2 = 0.0258
728
0170.00685.00114.00611.00242.00115.00258.0
−
++++++=cums
t
N
i
N
j
ji
cumulataNNN
xxxx
s−++
+−+−
=
∑ ∑= =
...
...)()(
21
1 1
2
2
2
1
1 2
Scum = 0.10 ppm Hg
Si noti che viene perso un grado di libertà per ognuno dei sette campioni. Poiché restano più di 20 gradi di libertà, comunque il valore calcolato di s può essere considerato una buona Approssimazione di σ, ovvero s → σ = 0.10 ppmHg.

28
LIVELLI DI FIDUCIA O DI CONFIDENZALIVELLI DI FIDUCIA O DI CONFIDENZA
Limiti di fiducia: definiscono un intervallo intorno a che con una certa probabilità contiene µ.
x
Intervallo di fiducia: è definito dai limiti di fiducia ed si riferisce alla probabilità che la vera media µ si trovi ad una certa distanza della media misurata x.
Livello di fiducia: fissa i limiti entro cui deve trovarsi il valore vero.
σµ zx ±=LF per
N
zx
σµ ±=LF per
Livelli di fiducia, % z50 0.6768 1.0080 1.2990 1.6495 1.9696 2.0099 2.5899.7 3.0099.9 3.29

29
INTERVALLO DI INTERVALLO DI FIDUCIAFIDUCIA QUANDO QUANDO σσσσσσσσ NON ENON E’’ NOTA: NOTA: la t di la t di StudentStudent
Un singolo insieme di misure replicate non solo deve fornire unamedia, ma anche consentire una stima della precisione. Il valore di s calcolato da un piccolo insieme di dati può essere piuttosto incerto.
Un singolo insieme di misure replicate non solo deve fornire unamediamedia, ma anche consentire una stima della precisione. Il valore di ss calcolato da un piccolo insieme di dati può essere piuttosto incerto.
quindi: i limiti di fiducia sono necessariamente più ampi!
s
xt
µ−=Parametro t: tiene in
considerazione la variabilità di s
N
tsx ±=µLivello di fiducia per la media x
di N misure replicate:
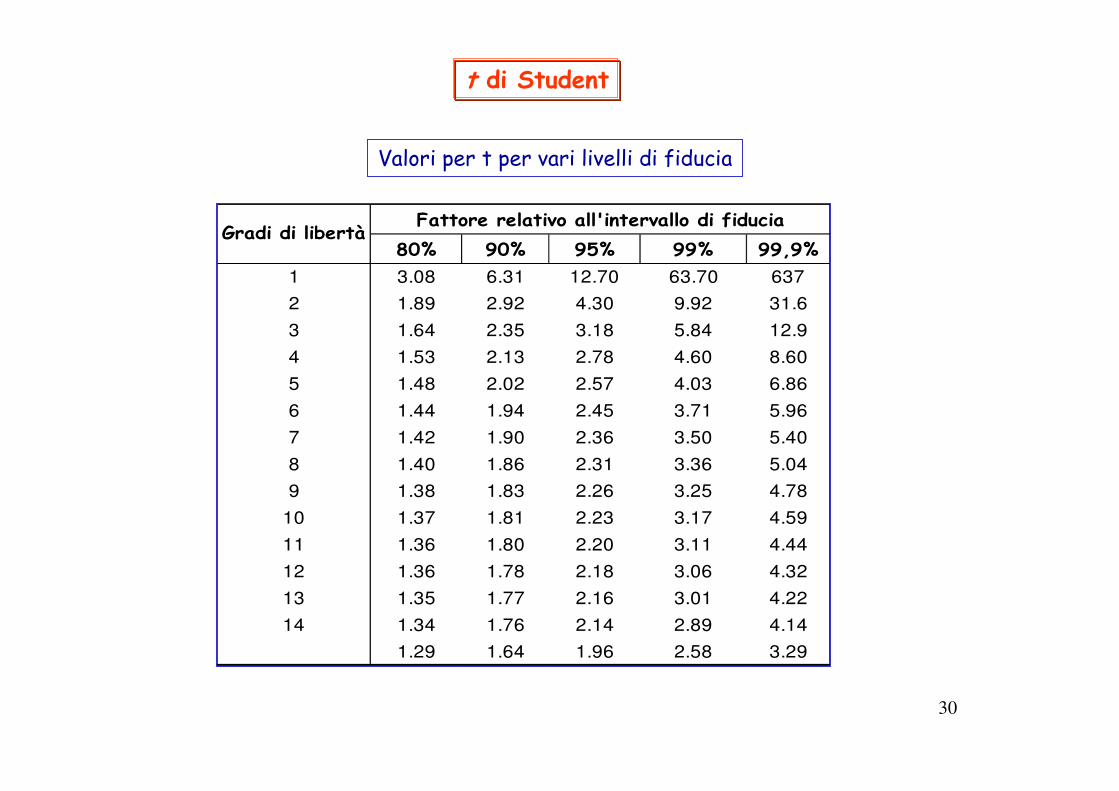
30
t di Student
Valori per t per vari livelli di fiducia
80% 90% 95% 99% 99,9%1 3.08 6.31 12.70 63.70 637
2 1.89 2.92 4.30 9.92 31.6
3 1.64 2.35 3.18 5.84 12.9
4 1.53 2.13 2.78 4.60 8.60
5 1.48 2.02 2.57 4.03 6.86
6 1.44 1.94 2.45 3.71 5.96
7 1.42 1.90 2.36 3.50 5.40
8 1.40 1.86 2.31 3.36 5.04
9 1.38 1.83 2.26 3.25 4.78
10 1.37 1.81 2.23 3.17 4.59
11 1.36 1.80 2.20 3.11 4.44
12 1.36 1.78 2.18 3.06 4.32
13 1.35 1.77 2.16 3.01 4.22
14 1.34 1.76 2.14 2.89 4.14
1.29 1.64 1.96 2.58 3.29
Fattore relativo all'intervallo di fiduciaGradi di libertà

31
Il test t viene utilizzato per confrontare fra loro due serie di misure, al fine di decidere se esse sono o non sono in accordo.
Statistica: fornisce una stima della probabilità che la differenza osservata sia dovuta semplicemente a errori casuali.
Decisione arbitraria: Si rifiuta l’ipotesi nulla nel caso in cui le probabilità di ottenere la differenza osservata a causa di errori casuali siano inferiore al 5%.
In base a questo criterio vi è il 95% di probabilità che le relative conclusioni siano vere.
Ipotesi nulla: i valori medi non differiscono tra loroIpotesi nulla: i valori medi non differiscono tra loro
tcalc > ttab : risultati sono diversi
tcalc < ttab : risultati sono statisticamente identici
tcalc > ttab : risultati sono diversi
tcalc < ttab : risultati sono statisticamente identici

32
Caso esempio 1: CONFRONTO TRA RISULTATO MISURATO E “VALORE VERO”Campione di riferimento (materiale certificato): carbone con 3.19% di zolfoRisultati ottenuti con un nuovo metodo analitico:
3.29%, 3.30 %, 3.22% e 3.23%media = 3.26% deviazione standard = 0.04
Il nuovo metodo è “valido”?
Calcoliamo il valore di t per questo set di misure e lo confrontiamo con il valore tabulato:
N
tsx ±=µ
4.3404.0
|19.326.3|=
−=calct
Ns
xxt verocalc
|""−=
Dalla tabella: al 95% con 3 gradi di libertà: ttab = 3.182tcalc > ttab il risultato ottenuto è diverso dal valore vero

33
Considera l’esempio della determinazione del mercurio nei pesci visto prima. Calcolare i
limiti di fiducia all’80% e al 95% per (a) il primo dato (1.80 ppm di Hg) e (b) il valore
medio (1.67 ppm di Hg) per il campione 1. Assumere in ogni caso che s →σ=0.1.
Dalla tabella t si ricava:t=1.29 per LF 80% t=1.96 per LF 95%
13.080.11
10.029.180.1%80 ±=±=
xLF
20.080.11
10.096.180.1%95 ±=±=
xLF
Osservando questi dati, si conclude che ci sono 80 probabilità su 100 che µ, la media della popolazione (ovvero, in assenza di errori sistematici, il valore vero) abbia un valore compreso tra 1.67 e 1.93 ppm di Hg. Inoltre, c’è una probabilità del 95% che essa abbia un valore compreso tra 1.60 e 2.00 ppm Hg.
Per una misura:Per una misura: Per tre misure:Per tre misure:
07.067.13
10.029.167.1%80 ±=±=
xLF
11.067.13
10.096.167.1%95 ±=±=
xLF
Cosi, la probabilità che la media della popolazione sia compresa tra 1.60 e 1.74 ppm di Hg è 80 su 100, mentre è di 95 su 100 che la media sia compresa tra 1.56 e 1.78 ppm.
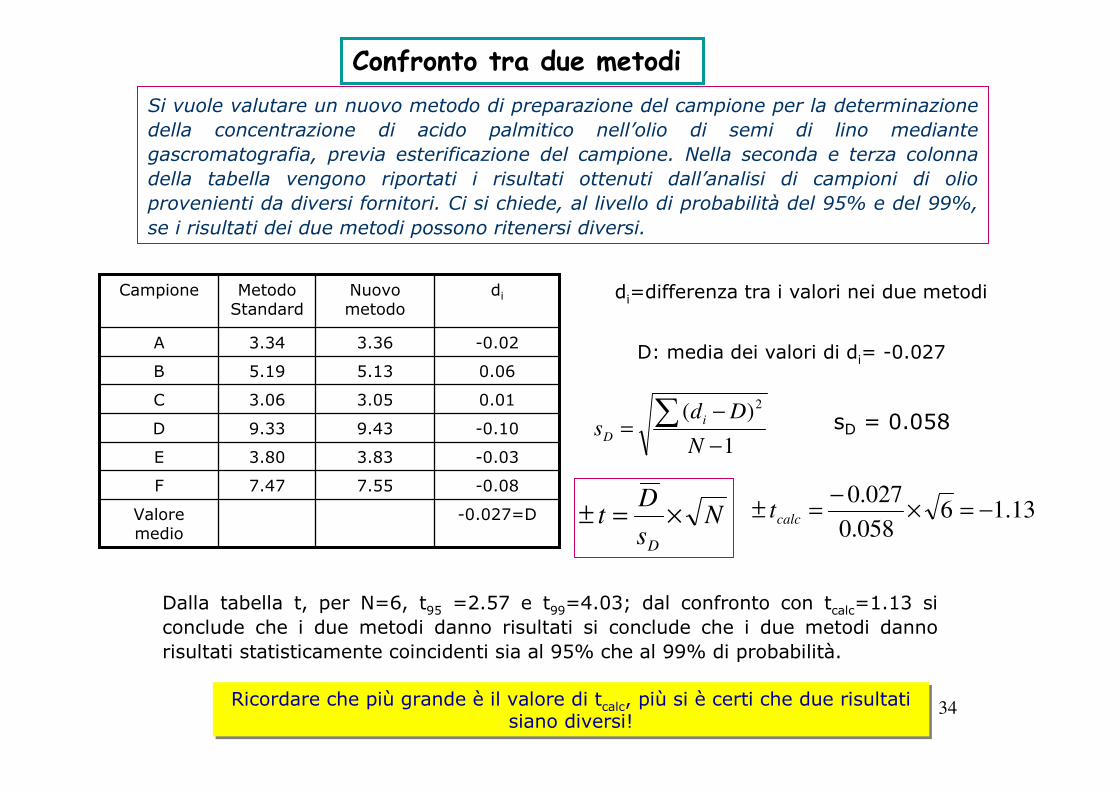
34
Confronto tra due metodi Si vuole valutare un nuovo metodo di preparazione del campione per la determinazione
della concentrazione di acido palmitico nell’olio di semi di lino mediante
gascromatografia, previa esterificazione del campione. Nella seconda e terza colonna
della tabella vengono riportati i risultati ottenuti dall’analisi di campioni di olio
provenienti da diversi fornitori. Ci si chiede, al livello di probabilità del 95% e del 99%,
se i risultati dei due metodi possono ritenersi diversi.
-0.027=DValore medio
-0.087.557.47F
-0.033.833.80E
-0.109.439.33D
0.013.053.06C
0.065.135.19B
-0.023.363.34A
diNuovo metodo
Metodo Standard
Campione di=differenza tra i valori nei due metodi
D: media dei valori di di= -0.027
sD = 0.0581
)( 2
−
−=∑
N
Dds
i
D
Ns
Dt
D
×=± 13.16058.0
027.0−=×
−=± calct
Dalla tabella t, per N=6, t95 =2.57 e t99=4.03; dal confronto con tcalc=1.13 si
conclude che i due metodi danno risultati si conclude che i due metodi danno
risultati statisticamente coincidenti sia al 95% che al 99% di probabilità.
Ricordare che più grande è il valore di tcalc, più si è certi che due risultati siano diversi!
Ricordare che più grande è il valore di tcalc, più si è certi che due risultati siano diversi!

35
E’ stato determinato il mercurio in sette pesci pescati nella Baia di Chesapeakecon un metodo basato sull’assorbimento di una radiazione da parte del mercurio gassoso elementare. Calcolare una stima accumulata della deviazione standard per il metodo utilizzato, basata sulle prime tre colonne di dati:
0.2196Somma dei
quadrati
28N°misure
0.01701.1301.11, 1.15, 1.22, 1.04
47
0.06852.4822.35, 2.44, 2.70,
2.48,2.44
56
0.01140.5700.57,0.58, 0.64, 0.49
45
0.06112.0182.06, 1.93, 2.12, 2.16, 1.89,1.95
640.02423.2403.13, 3.3523
0.01151.0150.96, 0.98, 1.02, 1.10
42
0.02581.6731.80,1.58,1.64
31
Somma quadrati
deviazione dalla media
[Hg]Media
(ppm)
[Hg]
(ppm)
N°campioni analizzati
Campione
Calcolo della media (4° colonna):
X= (1.80+1.58+1.64)/3= 1.673
Somma dei quadrati della deviazione della media:
(1.80-1.673)2+(1.80-1.,58)2+(1.80-1.64)2 = 0.0258
728
0170.00685.00114.00611.00242.00115.00258.0
−
++++++=cums
t
N
i
N
j
ji
cumulataNNN
xxxx
s−++
+−+−
=
∑ ∑= =
...
...)()(
21
1 1
2
2
2
1
1 2
Scum = 0.10 ppm Hg
Si noti che viene perso un grado di libertà per ognuno dei sette campioni. Poiché restano più di 20 gradi di libertà, comunque il valore calcolato di s può essere considerato una buona Approssimazione di σ, ovvero s → σ = 0.10 ppmHg.

36
Quante misure replicate del campione 1 dell’esempio del Hg sono
necessarie per ridurre l’intervallo a ± 0.07 ppm di Hg ad un livello di
fiducia del 95%?
L’intervallo di fiducia (IF) è dato dal secondo termine sulla destra
dell’equazione:
N
zIF
σ±=
quindi:
NN
zIF
10.096.107.0
×±=±==
σ
8.7=N
Con otto misure e di conseguenza una probabilità leggermente
superiore al 95% si potrebbe ottenere una media della popolazione
compresa nell’intervallo ±0.07 ppm rispetto alla media
sperimentale.

37
Un chimico ha ottenuto i seguenti risultati relativi al contenuto di alcol
in un campione di sangue: 0.084%, 0.089% e 0.079%. Calcolare i
limiti di fiducia della media al 95% assumendo che:
a) Non si ha alcuna conoscenza aggiuntiva sulla precisione del
metodo;
b) Sulla base di esperienza precedenti, si sa che s→σ→σ→σ→σ= 0.005% di alcol.
a)
1
/)( 22
−
−=∑ ∑
N
Nxxs
ii %0050.0=s C2H5OH
In questo caso, la media è 0.252/3=0.084. Dalla tabella t=4.30 per due gradi di libertàe al livello di fiducia del 95%. Cosi:
3
0050.030.4084.0%95
×±=±=
N
tsxLF =0.084±0.012% C2H5OH
b)Poiché è disponibile un valore accettabile di σ:
3
0050.096.1084.0%95
×±=±=
N
tsxLF =0.084±0.006% C2H5OH
Si nota che l’intervallo di fiducia decresce notevolmente quando σσσσ è nota.
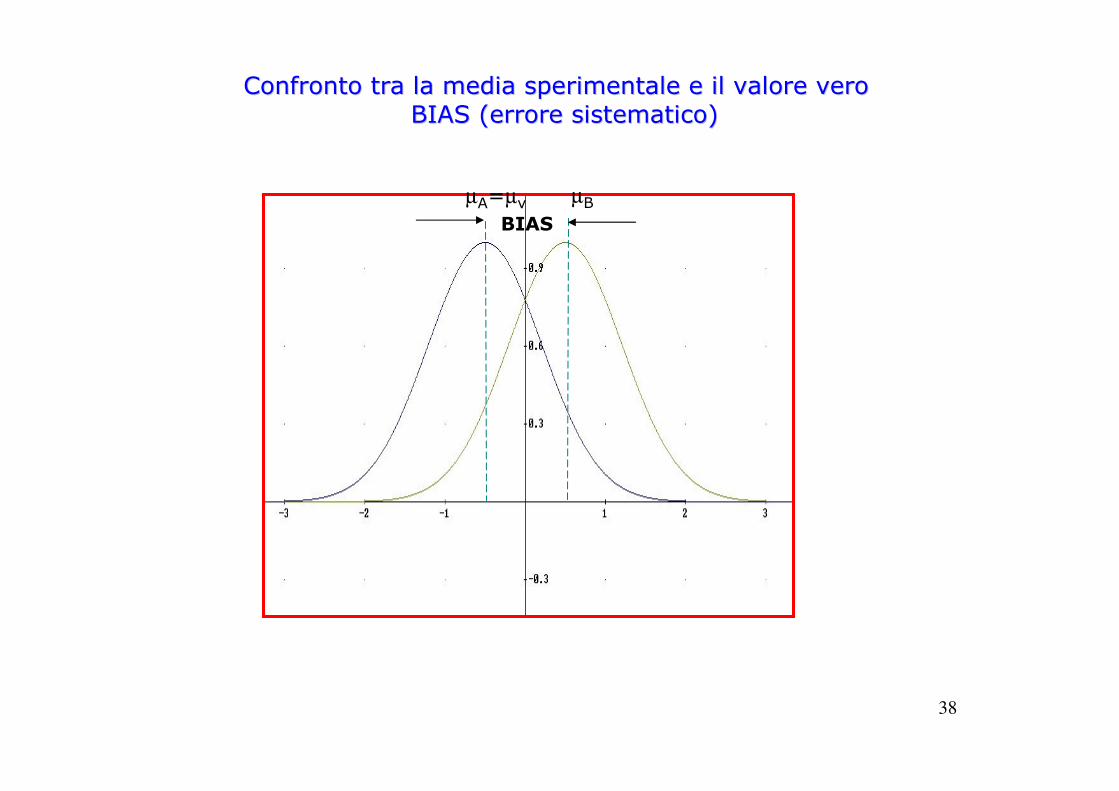
38
Confronto tra la media sperimentale e il valore veroConfronto tra la media sperimentale e il valore veroBIAS (errore sistematico)BIAS (errore sistematico)
BIAS
µA=µv µB

39
ESEMPIOESEMPIO– Si desidera valutare l’esattezza di un nuovo metodo di analisi
elettrochimica del cobalto nelle ceneri di inceneritori comunali. Allo scopo, un
operatore analizza ripetutamente il materiale di riferimento certificato CRM176
(cenere di inceneritore cittadino contenente la concentrazione di analita CCo =
30,9 mg/kg). I risultati delle 11 analisi sono i seguenti :
CCo (mg/kg) : 28,9; 29,8; 29,9; 30,6; 28,5; 31,2; 32,1; 30,6; 30,9; 31,7; 30,0
Il risultato del bianco non è significativamente diverso da zero. Valutare
l’esattezza del risultato fornito dal nuovo metodo (P = 95%).
Calcolo del valore medio e stima della deviazione standard:
CmCo = 30,382 mg/kg s = 1,103 mg/kg t1-α/2,10 = 2,228
Intervallo di fiducia:
Risultato: Cexp = 30,38 ± 0,74 mg/kg (α = 0,05; ν = 10)
L’intervallo di fiducia comprende il valore certificato, 30,9 mg/kg, e quindi non
si ha evidenza di bias nei limiti del livello di fiducia prescelto.
741.011
st 10,2/1 =⋅α−

40
E’ stata verificata una nuova procedura per la rapida determinazione dello zolfo nei cheroseni; è noto è noto dal suo metodo di preparazione cheil campione analizzato contiene lo 0.123% (xv=0.123) di S. I risultati sono stati: %S: 0.112 0.118 0.115 0.119Sulla base di questi dati è possibile affermare che il metodo presenta un errore sistematico?
x = 0.464/4 = 0.116 % S
=− vxx 0.116-0.123= -0.007%S
= 0.112+0.118+0.115+0.119= 0.464∑ ix
= 0.012544+0.013924+0.013225+0.014161=0.0538542
∑ ix
0032.014
4/)464.0(053854.0 2
=−
−=s
Per LF 95% e tre gradi di libertà, t=3.18. Quindi:0051.0
4
0032.018.3
4±=
×=
ts
Ci si può aspettare che una media sperimentale presenti deviazioni di ±0.0051 o maggiori non più frequentemente di 5 volte su 100.Cosi, se concludiamo che -0.007 è una differenza significativa e che un erroresitematico è presente, ci sbaglieremo , in media, meno di 5 volte su 100.
=− vxx

41
Confronto tra due medie sperimentali
21
2121
NN
NNtsxx cumulata
+±=−
E’ stato analizzato il contenuto di alcool di due botti di vino, per determinare se essi avessero origine diversa. Sulla base di sei analisi, è stato stabilito che il contenuto medio in etanolo della prima botte è 12,61%. Per la seconda botte, la media di quattro analisi èrisultata 12.53% di alcool. Le dieci analisi hanno prodotto un valore cumulato di s pari allo 0.070%. Sulla base di questi dati, è possibile affermare che c’è una differenza tra i due vini?
In questo caso è possibile impiegare l’equazione sopra, utilizzando t per otto gradi di libertà (10-2). Al livello di fiducia del 95%:
%10.046
46070.031.2
21
21 ±=×
+×±=
+±
NN
NNts
La differenza osservata è: %08.053.1261.1221 =−=− xx
Con una media di 5 volte su 100 l’errore casuale causerà una differenza pari a 0.10%. Al livello di fiducia del 95% dunque, non è stata
dimostrata alcuna differenza tra i contenuti di alcool nei due vini!

42
DETERMINAZIONE DI ERRORI GROSSOLANIDETERMINAZIONE DI ERRORI GROSSOLANI
Gli outliers o dati anomali sono il risultato di errori grossolani
Quando un insieme di dati contiene un risultato che sembra differire eccessivamente dalla media (outlier), bisogna adottare dei criteri opportuni per decidere se scartalo o meno.
Non esiste una regola universale che consenta di decidere di scartare o di accettare un outlier!
Il test Q: wxxQ nqsper /|| −=
Xq : risultato dubbio
xn: risultato più vicino al risultato dubbio
w : dispersione (differenza tra il valore piùgrande e quello più piccolo dell’insieme)
Confrontare Qsper con Qcrit
dalla tabella:
Se Qsper > Qcrit: SCARTARE CON L’INTERVALLO DI FIDUCIA INDICATO NELLA TABELLA.
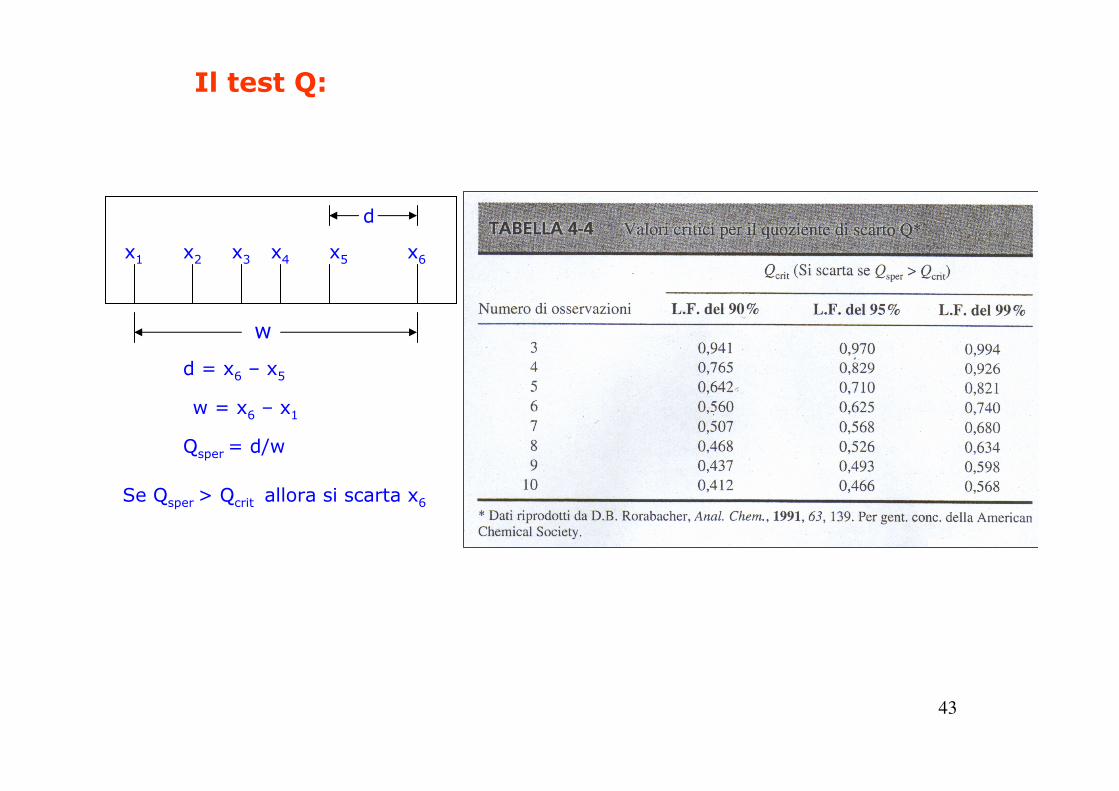
43
x1 x6x5x4x3x2
d
w
d = x6 – x5
w = x6 – x1
Qsper = d/w
Se Qsper > Qcrit allora si scarta x6
Il test Q:

44
CIFRE DI MERITO PER VALUTARE LA PRECISIONE DEI METODI ANALITICICIFRE DI MERITO PER VALUTARE LA PRECISIONE DEI METODI ANALITICI
Termini Definizione
xi = valore numerico dell’iesima misura = media di N misureN
x
x
N
i
i∑== 1
Varianza s2
Deviazione standard assoluta
1
)(1
2
−
−
=
∑=
N
xx
s
N
i
i
Deviazione standard relativa (RSD) x
sRSD =
Deviazione standard della media (Sm) N
ssm =
Coefficiente di variazione (CV) %100×=x
sCV

45
Applicazione della statistica alle misure effettuate nell’analisi chimica
METODO DI TARATURA
Curva di taratura
Metodo delle aggiunte standard
Standard interno
46
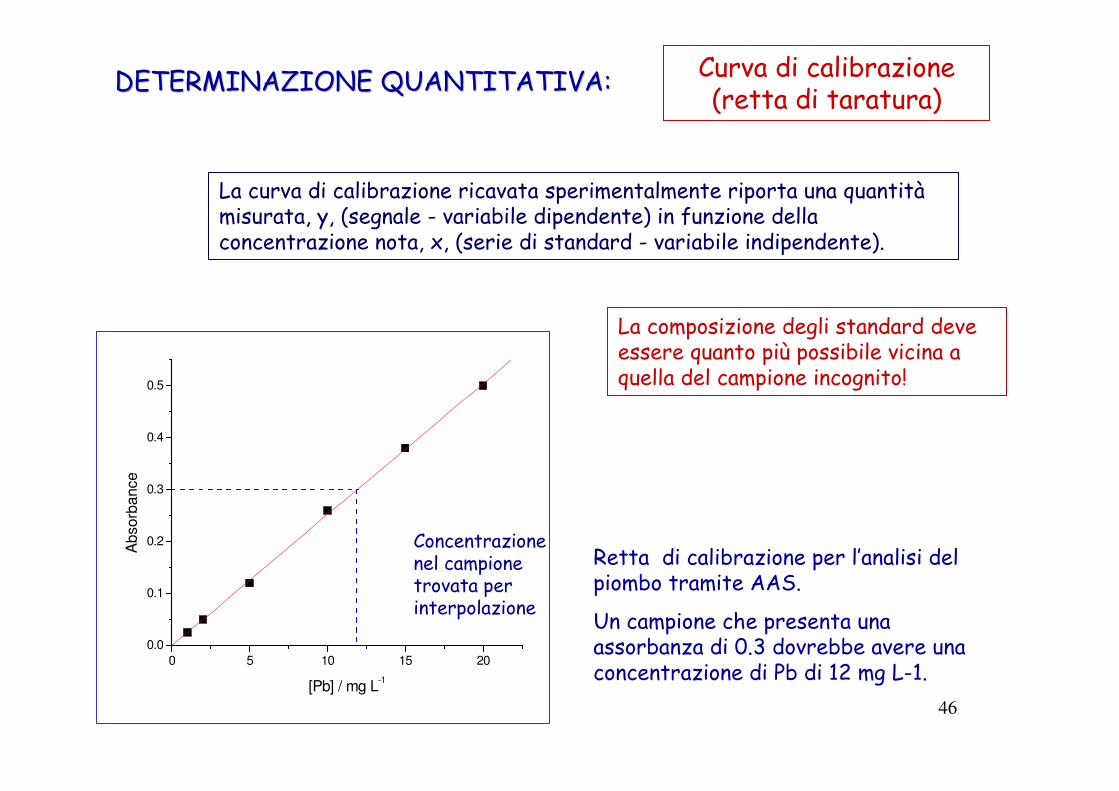
DETERMINAZIONE QUANTITATIVA:DETERMINAZIONE QUANTITATIVA: Curva di calibrazione(retta di taratura)
Retta di calibrazione per l’analisi del piombo tramite AAS.
Un campione che presenta una assorbanza di 0.3 dovrebbe avere una concentrazione di Pb di 12 mg L-1.
0 5 10 15 20
0.0
0.1
0.2
0.3
0.4
0.5
Absorb
ance
[Pb] / mg L-1
Concentrazione nel campione trovata per interpolazione
La curva di calibrazione ricavata sperimentalmente riporta una quantitàmisurata, y, (segnale - variabile dipendente) in funzione della concentrazione nota, x, (serie di standard - variabile indipendente).
La composizione degli standard deve essere quanto più possibile vicina a quella del campione incognito!

47
Retta di taratura
Determinare l’equazione di regressione della retta di taratura.
Controllare che il coefficiente di determinazione (R2) sia il piùpossibile vicino a 1. Controllare ed eventualmente scartare i punti aberranti.
Controllare che l’intercetta sia significativamente diversa da zero. Se questo non si verifica, indagare sulle cause.
Determinare le concentrazioni incognite, magari su campioni replicati più volte, con il relativo intervallo di fiducia.
Riutilizzare la retta in più sessioni analitiche, fino a quando i reagenti con i quali è stata determinata non sono terminati. Per sicurezza si possono preparare ogni volta uno o più standard di controllo, verificando che i segnali misurati cadano entro l’intervallo di fiducia della misura attesa.

48
Sensibilità di un metodo
La sensibilità (S) di un metodo è data dal rapporto tra la variazione del segnale (dy) in funzione della corrispondente variazione della concentrazione (dx), che esprime la variazione del segnale per ogni variazione di concentrazione unitaria
dx
dyS =
Corrisponde alla pendenza della retta di calibrazione

49
Il metodo dei minimi quadrati per la realizzazione di curve Il metodo dei minimi quadrati per la realizzazione di curve di calibrazionedi calibrazione
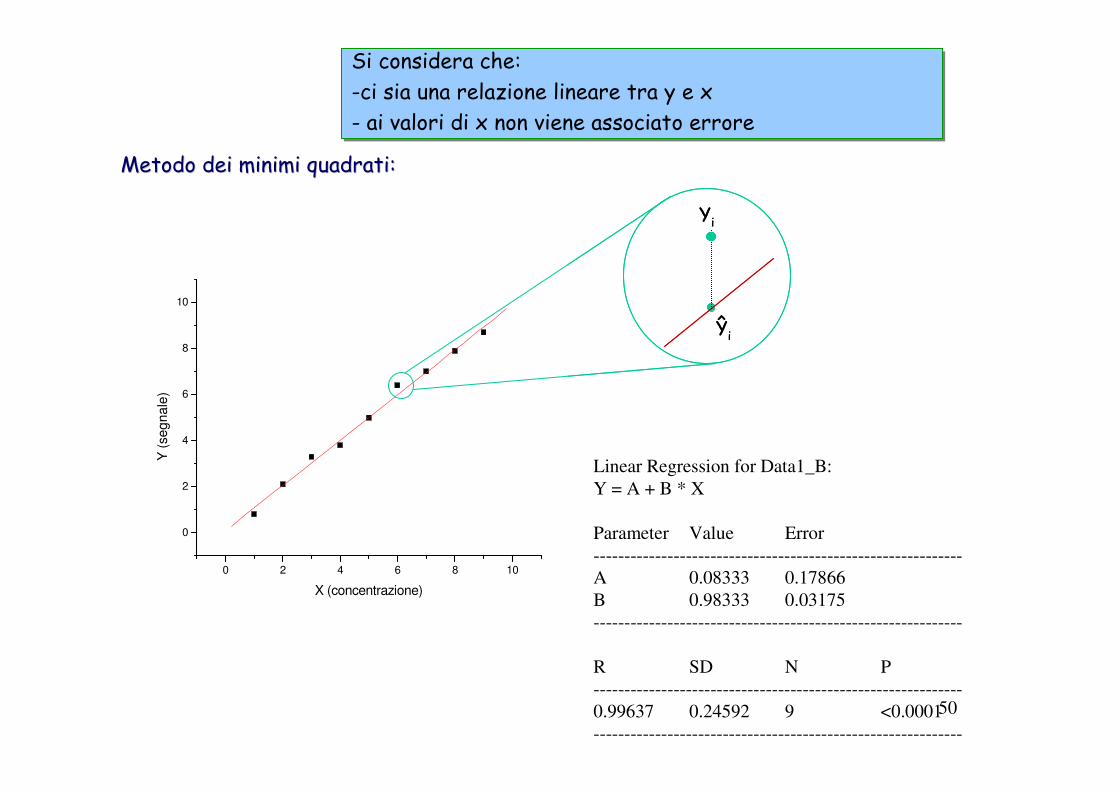
A causa degli errori indeterminati associati al processo di misurazione, non tutti i dati si trovano esattamente sulla retta.
Per cercare di derivare la migliore retta che interpoli i punti si usa la tecnica statistica chiamata analisi di regressione - essa consente di ottenere tale retta in maniera obiettiva e di specificare le incertezza associate. Useremo il metodo metodo dei minimi quadrati.dei minimi quadrati.
La retta di calibrazione viene definita algebricamente come:
Yi = mXi + bdove: Yi è il risultato analitico
Xi è la concentrazione dell’analita corrispondente a Yim è la pendenza della retta (ossia la sensibilità del metodo)b è una costante chiamata intercetta, che rappresenta il valore di Yi quando Xi = 0. (non considerare questo valore contribuisce all’errore sistematico del metodo).
50
0 2 4 6 8 10
0
2
4
6
8
10
Y (
se
gn
ale
)
X (concentrazione)
Ŷi
Yi
Metodo dei minimi quadrati:Metodo dei minimi quadrati:
Linear Regression for Data1_B:
Y = A + B * X
Parameter Value Error
------------------------------------------------------------
A 0.08333 0.17866
B 0.98333 0.03175
------------------------------------------------------------
R SD N P
------------------------------------------------------------
0.99637 0.24592 9 <0.0001
------------------------------------------------------------
0 2 4 6 8 10
0
2
4
6
8
10
Y (
se
gn
ale
)
X (concentrazione)
Ŷi
Yi
Si considera che: -ci sia una relazione lineare tra y e x- ai valori di x non viene associato errore
Si considera che: -ci sia una relazione lineare tra y e x- ai valori di x non viene associato errore
51
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,4 1,6 1,8
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
se
gn
ale
Conc analita
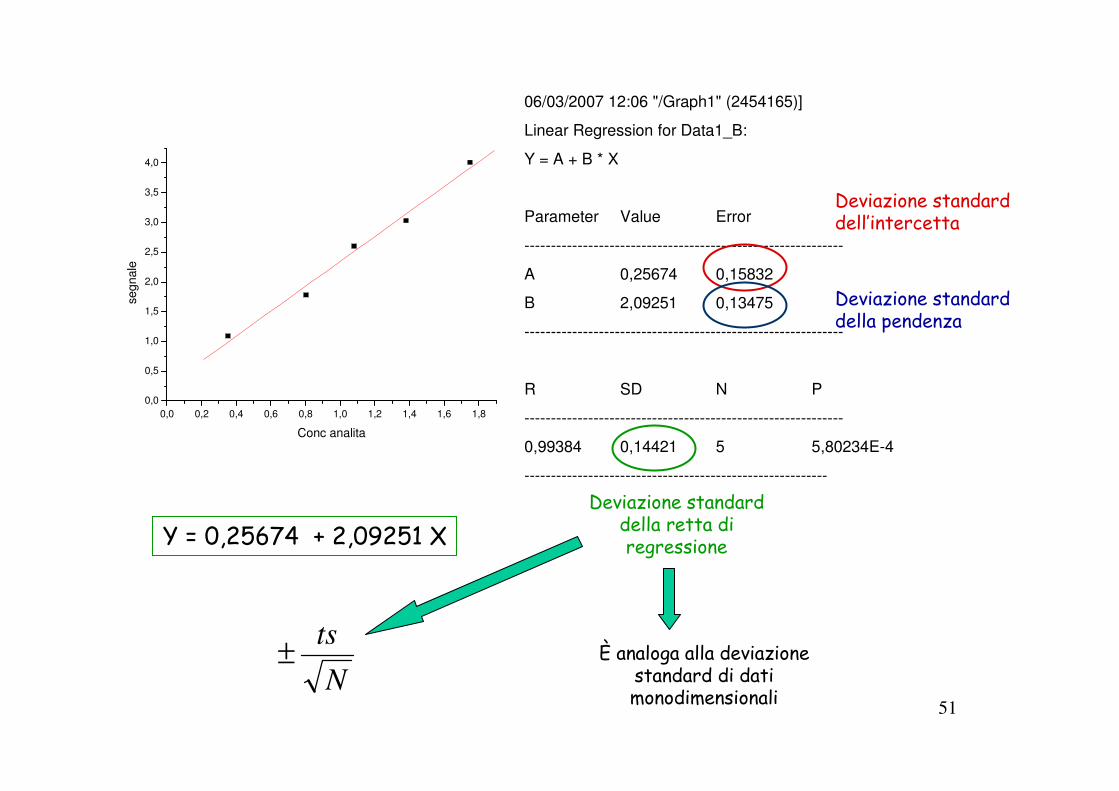
06/03/2007 12:06 "/Graph1" (2454165)]
Linear Regression for Data1_B:
Y = A + B * X
Parameter Value Error
------------------------------------------------------------
A 0,25674 0,15832
B 2,09251 0,13475
------------------------------------------------------------
R SD N P
------------------------------------------------------------
0,99384 0,14421 5 5,80234E-4
---------------------------------------------------------
Y = 0,25674 + 2,09251 X
Deviazione standard dell’intercetta
Deviazione standard della pendenza
Deviazione standard della retta di regressione
È analoga alla deviazione standard di dati monodimensionali
N
ts±
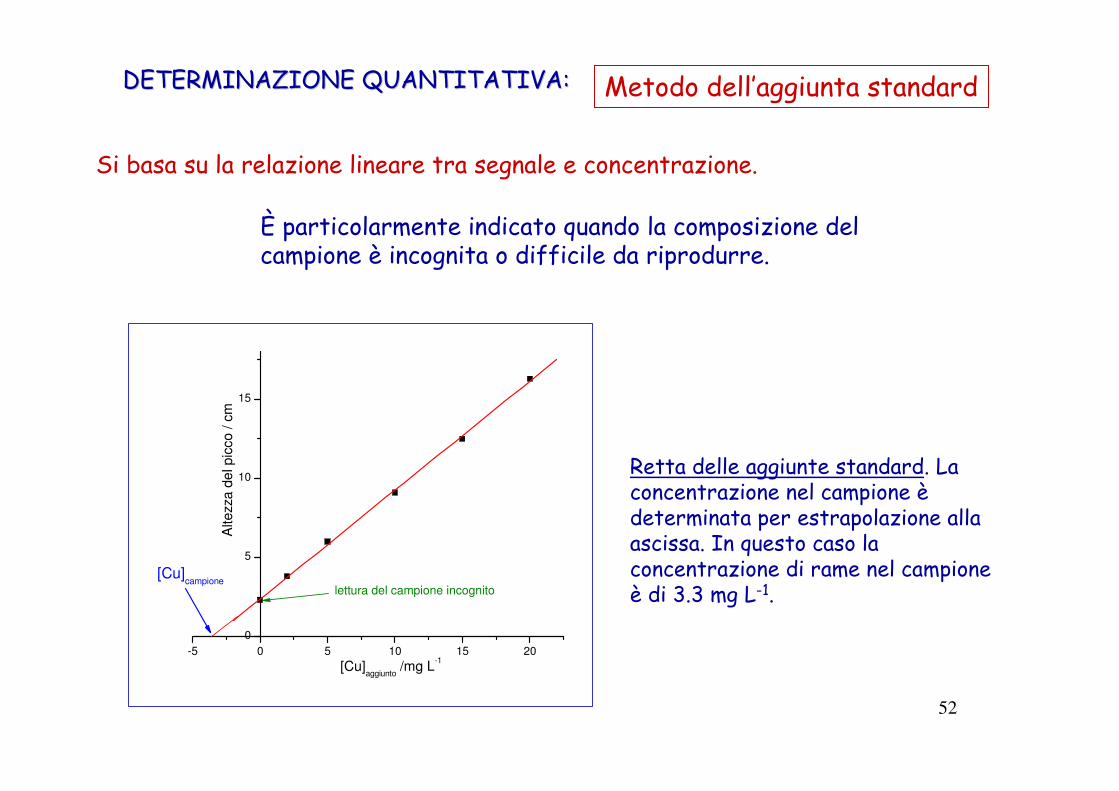
52
Retta delle aggiunte standard. La concentrazione nel campione èdeterminata per estrapolazione alla ascissa. In questo caso la concentrazione di rame nel campione è di 3.3 mg L-1.
-5 0 5 10 15 20
0
5
10
15
Altezza d
el pic
co / c
m
[Cu]aggiunto
/mg L-1
[Cu]campione
lettura del campione incognito
Si basa su la relazione lineare tra segnale e concentrazione.
È particolarmente indicato quando la composizione del campione è incognita o difficile da riprodurre.
DETERMINAZIONE QUANTITATIVA:DETERMINAZIONE QUANTITATIVA: Metodo dell’aggiunta standard
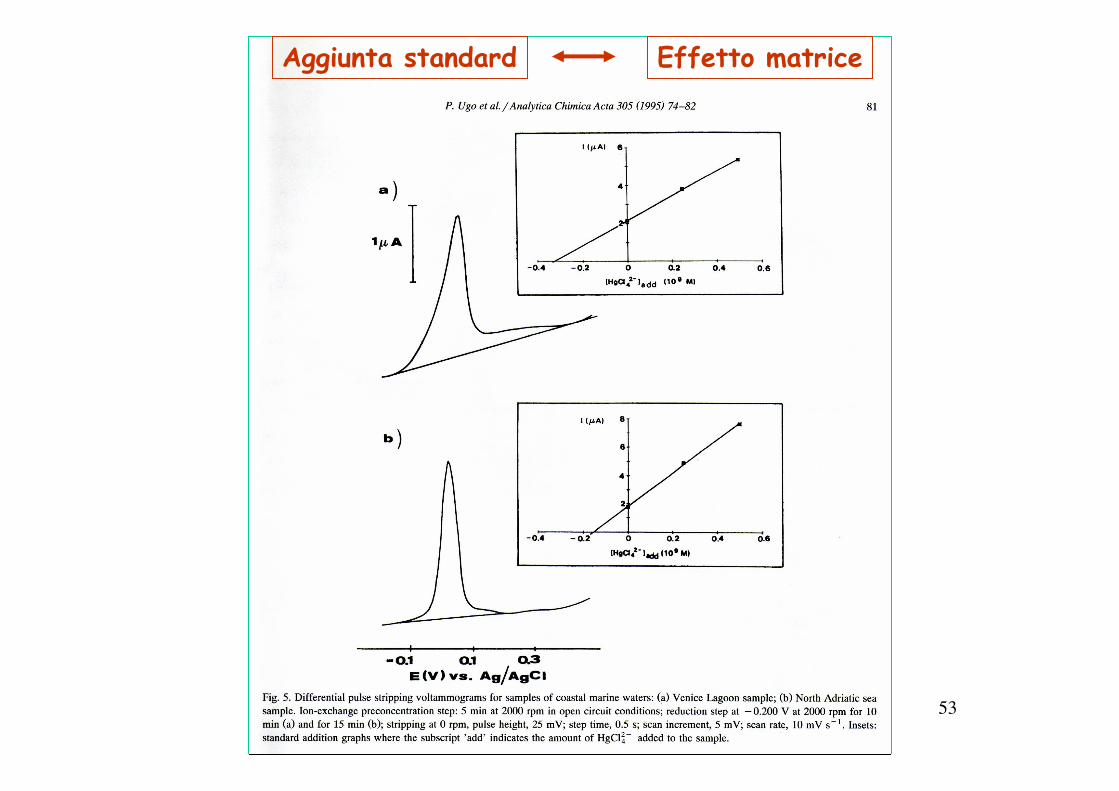
53
Aggiunta standard Effetto matrice

54
Metodo dell’aggiunta standard o multipla
Determinare l’equazione di regressione della retta dei risultati delle aggiunte
Controllare che il coefficiente di determinazione (R2) sia il piùpossibile vicino a 1. Controllare ed eventualmente scartare i punti aberranti.
Controllare che l’intercetta sia significativamente diversa da zero.
Se l’intercetta non è diversa da zero, esprimere un responso negativo (analita assente o inferiore ai limiti di rivelabilità o di quantificazione).
Determinare la concentrazione incognita ponendo y (segnale) = 0 e calcolare l’intervallo di fiducia.
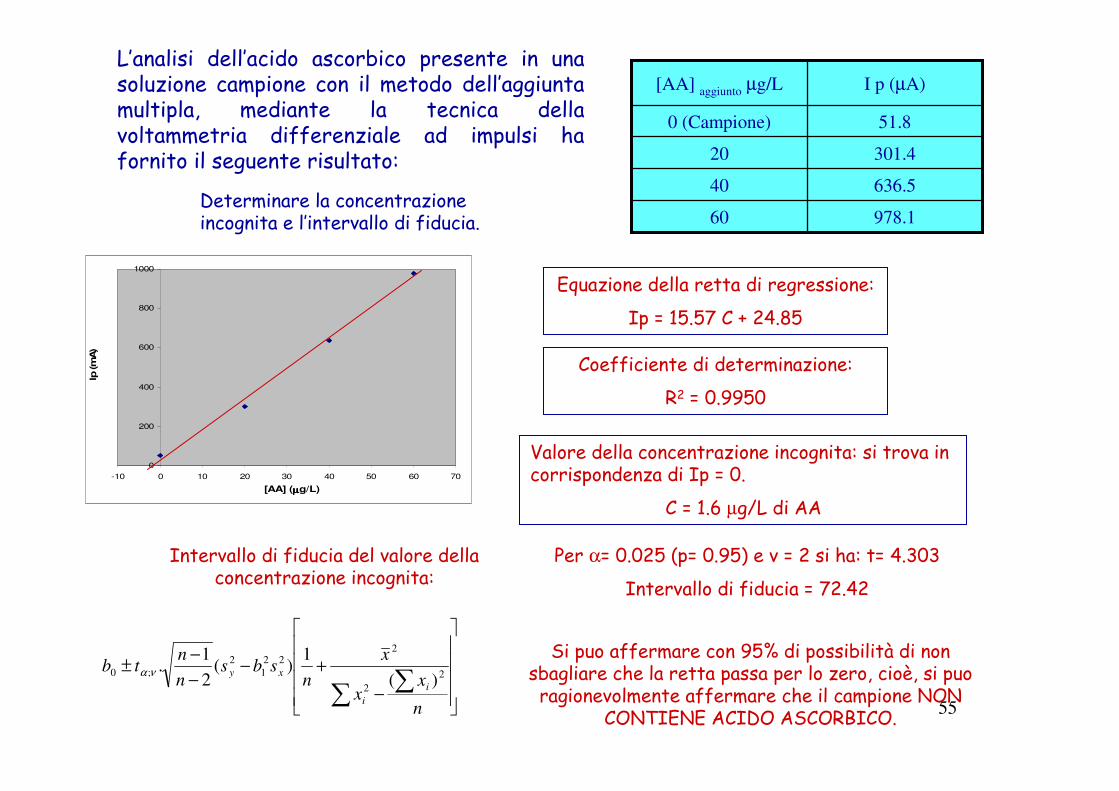
55
L’analisi dell’acido ascorbico presente in una soluzione campione con il metodo dell’aggiunta multipla, mediante la tecnica della voltammetria differenziale ad impulsi ha fornito il seguente risultato:
978.160
636.540
301.420
51.80 (Campione)
I p (µA)[AA] aggiunto µg/L
Determinare la concentrazione incognita e l’intervallo di fiducia.
0
200
400
600
800
1000
-10 0 10 20 30 40 50 60 70
[AA] (µµµµg/L)
Ip (m
A)
Equazione della retta di regressione:
Ip = 15.57 C + 24.85
Coefficiente di determinazione:
R2 = 0.9950
Valore della concentrazione incognita: si trova in corrispondenza di Ip = 0.
C = 1.6 µg/L di AA
Intervallo di fiducia del valore della concentrazione incognita:
−
+−−
−±
∑ ∑n
xx
x
nsbs
n
ntb
i
i
xy 2
2
222
1
2
;0)(
1)(
2
1.να
Per α= 0.025 (p= 0.95) e v = 2 si ha: t= 4.303
Intervallo di fiducia = 72.42
Si puo affermare con 95% di possibilità di non sbagliare che la retta passa per lo zero, cioè, si puo
ragionevolmente affermare che il campione NON CONTIENE ACIDO ASCORBICO.
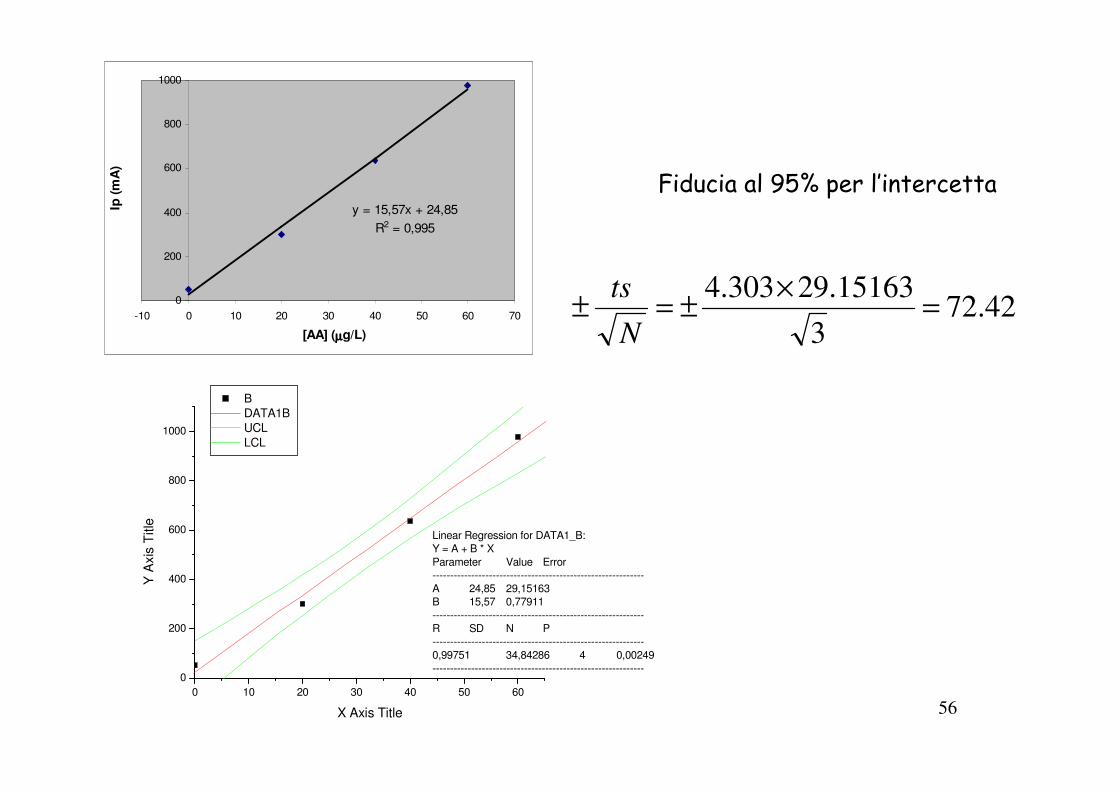
56
y = 15,57x + 24,85
R2 = 0,995
0
200
400
600
800
1000
-10 0 10 20 30 40 50 60 70
[AA] (µµµµg/L)
Ip (
mA
)
0 10 20 30 40 50 60
0
200
400
600
800
1000
Linear Regression for DATA1_B:
Y = A + B * X
Parameter Value Error
------------------------------------------------------------
A 24,85 29,15163
B 15,57 0,77911
------------------------------------------------------------
R SD N P
------------------------------------------------------------
0,99751 34,84286 4 0,00249
------------------------------------------------------------
Y A
xis
Title
X Axis Title
B
DATA1B
UCL
LCL
42.723
15163.29303.4=
×±=±
N
ts
Fiducia al 95% per l’intercetta

57
RANGE DINAMICO E LINEARE
Il range è l’intervallo di concentrazione esplorato nel corso delle misurazioni.
Il range dinamico è l'intervallo di concentrazione nel quale il segnale varia con la concentrazione: i limiti inferiore e superiore del range dinamico corrispondono, rispettivamente, al limite di rivelabilità ed alla più alta concentrazione alla quale un incremento di concentrazione produce ancora un incremento di segnale.
Il range lineare esprime l'intervallo di concentrazione nel quale il segnale varia linearmente con la concentrazione.
La costruzione del diagramma di calibrazione implica l’adozione di un metodo di regressione. Quello più generalmente adottato è il metodo di regressione lineare ordinaria dei minimi quadrati (OLLSR).
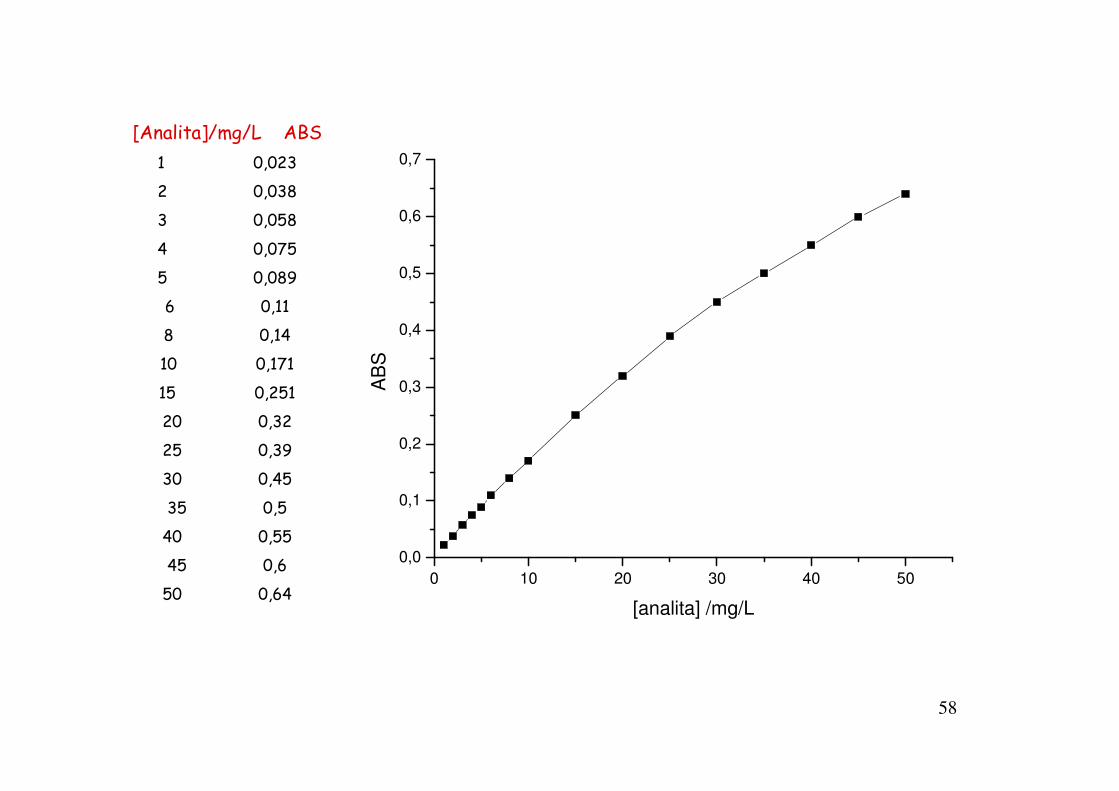
58
0 10 20 30 40 50
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
AB
S
[analita] /mg/L
[Analita]/mg/L ABS1 0,023
2 0,038
3 0,058
4 0,075
5 0,089
6 0,11
8 0,14
10 0,171
15 0,251
20 0,32
25 0,39
30 0,45
35 0,5
40 0,55
45 0,6
50 0,64

59
DETERMINAZIONE QUANTITATIVA:DETERMINAZIONE QUANTITATIVA:
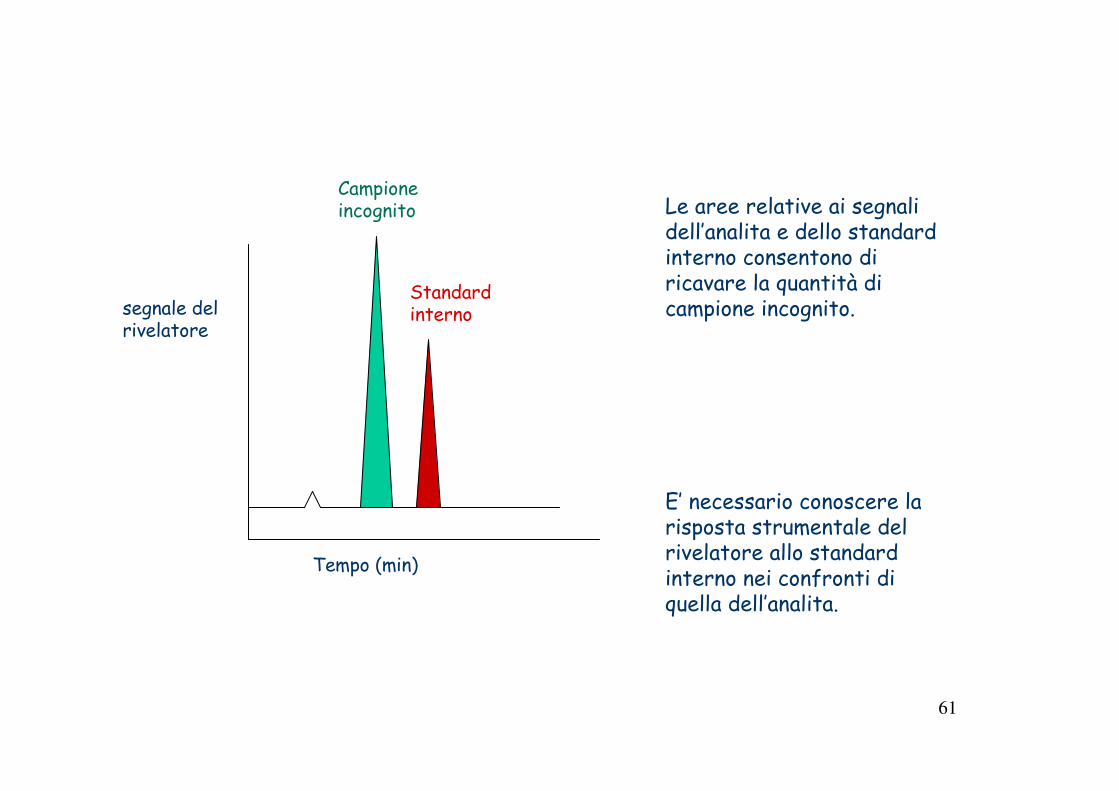
Metodo dello standard interno
Lo standard interno è una specie chimica diversa dall’analita, che viene aggiunta in quantità nota al campione incognito.La quantificazione viene fatta dal confronto tra il segnale dell’analita con il segnale dello standard interno
Quando si usa:� Sono piu usati in cromatografia.� quando la quantità di campione varia da una prova all’altra in
condizioni difficili da controllare;� Per aiutare a identificare un evento di perdita del campione
durante il processo analitico� Lo standard interno deve avere proprietà note e stabili.

60
Metodo dello standard interno: è il metodo di quantificazione più usato nell’analisi gascromatografica, in quanto consente di ottenere risultati più accurati ed affidabili, soprattutto se applicato al metodo della retta di taratura. In pratica, non si fa riferimento al picco dell’analita, ma al rapporto dell’area di questo e l’area di un componente (lo standard interno), aggiunto in quantità nota alla miscela da analizzare. In questo modo si prepara la retta di taratura ovviando a problemi tecnici quali la riproducibilità del sistema di iniezione ed eventuali derive della sensibilità del rivelatore.
La sostanza scelta come standard interno deve obbedire ad una serie di requisiti:
• non essere presente nel campione da analizzare;• Essere stabile termicamente;• Fornire un picco cromatografico ben risolto (non sovrapposto) da quello
degli altri componenti • Avere un tempo di ritenzione simile a quello dei componenti da
determinare;• Essere strutturalmente simile a questi ( e quindi avere un fattore di
risposta simile)• Essere sufficientemente pura e non reagire con i componenti del campione.
61
Tempo (min)
segnale del rivelatore
Campione incognito
Standard interno
Le aree relative ai segnali dell’analita e dello standard interno consentono di ricavare la quantità di campione incognito.
E’ necessario conoscere la risposta strumentale del rivelatore allo standard interno nei confronti di quella dell’analita.
62
Segnale misurato
Valore medio del segnale del
fondo sf
zero
Segnale al Limite di rivelabilità:
sf+3σb
10σb
Segnale al Limite di quantificazione:
sf+10σb
Segnale dovuto al solo
analita
Zona di rivelazione
Zona di quantificazione
Probabilità di ottenere il segnale
3σb
63
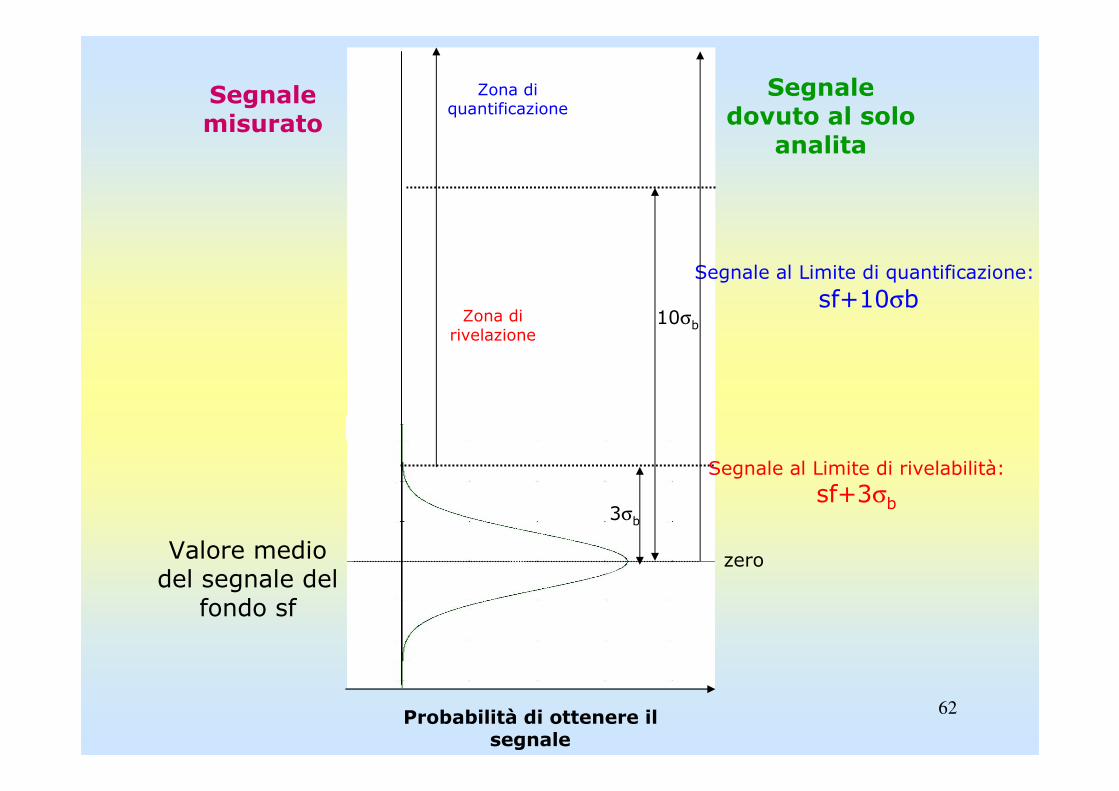
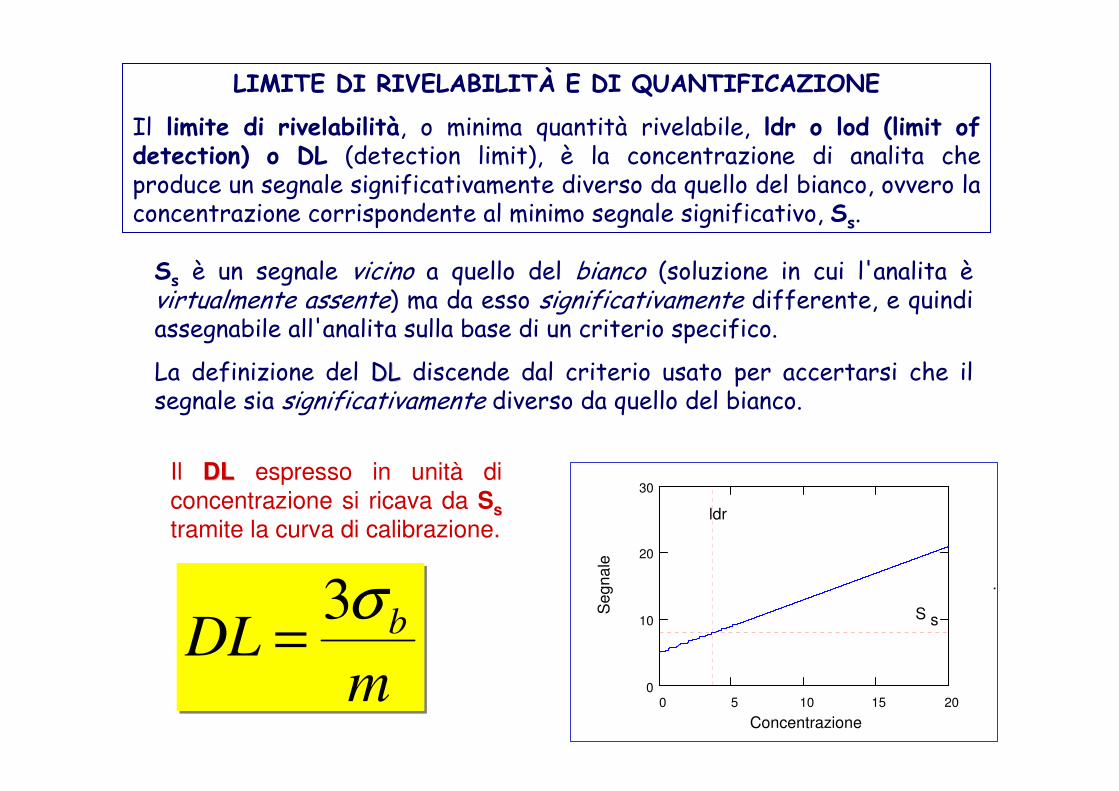
LIMITE DI RIVELABILITÀ E DI QUANTIFICAZIONE
Il limite di rivelabilità, o minima quantità rivelabile, ldr o lod (limit of detection) o DL (detection limit), è la concentrazione di analita che produce un segnale significativamente diverso da quello del bianco, ovvero la concentrazione corrispondente al minimo segnale significativo, Ss.
Ss è un segnale vicino a quello del bianco (soluzione in cui l'analita èvirtualmente assente) ma da esso significativamente differente, e quindi assegnabile all'analita sulla base di un criterio specifico.
La definizione del DLDL discende dal criterio usato per accertarsi che il segnale sia significativamente diverso da quello del bianco.
S s
ldr
0 5 10 15 200
10
20
30
Concentrazione
Se
gn
ale
.
Il DLDL espresso in unità di
concentrazione si ricava da Ss
tramite la curva di calibrazione.
mDL bσ3
=
64
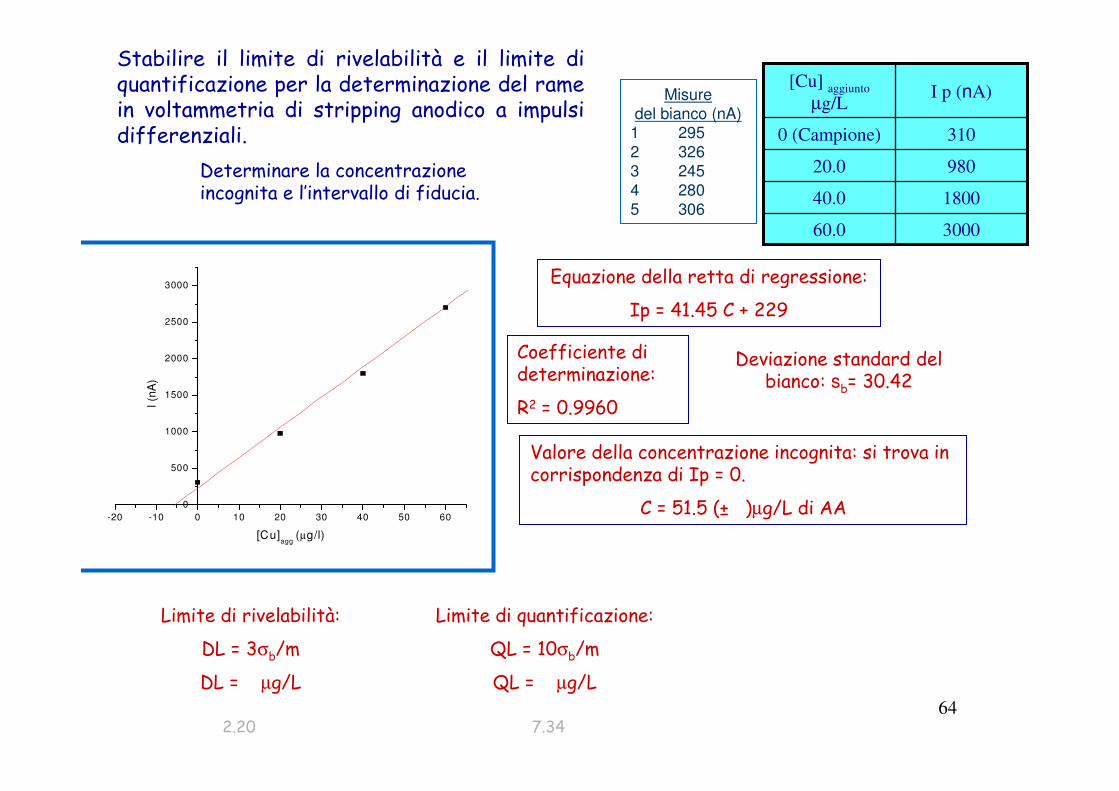
Stabilire il limite di rivelabilità e il limite di quantificazione per la determinazione del rame in voltammetria di stripping anodico a impulsi differenziali.
300060.0
180040.0
98020.0
3100 (Campione)
I p (nA)[Cu] aggiunto
µg/L
Determinare la concentrazione incognita e l’intervallo di fiducia.
Equazione della retta di regressione:
Ip = 41.45 C + 229
Coefficiente di determinazione:
R2 = 0.9960
Valore della concentrazione incognita: si trova in corrispondenza di Ip = 0.
C = 51.5 (± )µg/L di AA
Limite di rivelabilità:
DL = 3σb/m
DL = µg/L
Misure
del bianco (nA)1 295
2 326
3 245
4 280
5 306
-20 -10 0 10 20 30 40 50 60
0
500
1000
1500
2000
2500
3000
I (n
A)
[Cu]agg
(µg/l)
Deviazione standard del bianco: sb= 30.42
Limite di quantificazione:
QL = 10σb/m
QL = µg/L
2.20 7.34
65
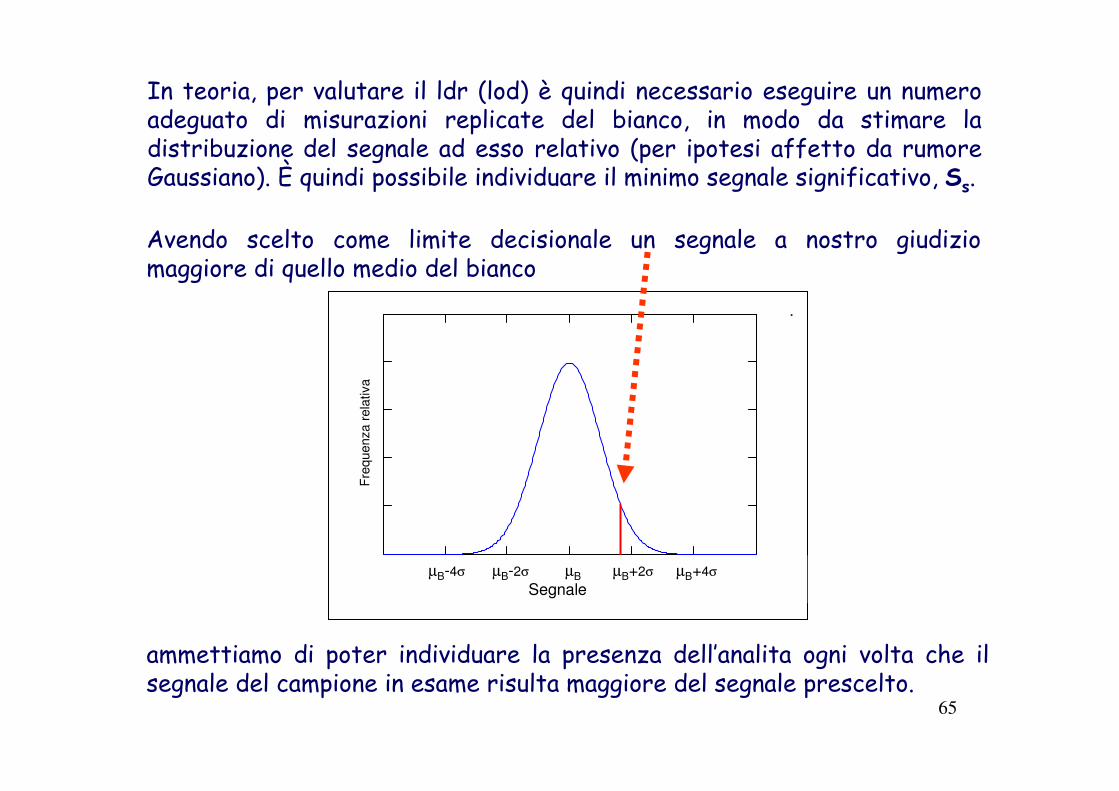
Avendo scelto come limite decisionale un segnale a nostro giudizio maggiore di quello medio del bianco
In teoria, per valutare il ldr (lod) è quindi necessario eseguire un numero adeguato di misurazioni replicate del bianco, in modo da stimare la distribuzione del segnale ad esso relativo (per ipotesi affetto da rumore Gaussiano). È quindi possibile individuare il minimo segnale significativo, Ss.
6 4 2 0 2 4 6
Segnale
Fre
quenza r
ela
tiva
.
µB-4σ µB-2σ µB µB+2σ µB+4σ
Segnale
ammettiamo di poter individuare la presenza dell’analita ogni volta che il segnale del campione in esame risulta maggiore del segnale prescelto.
66
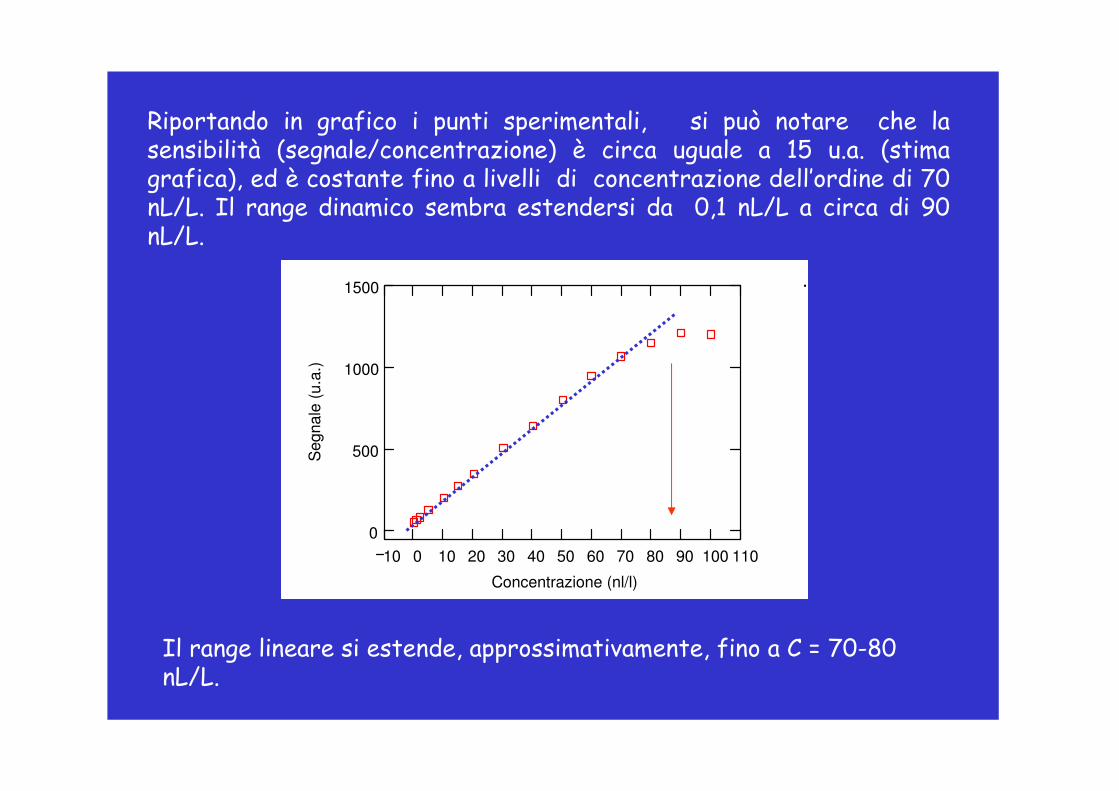
Riportando in grafico i punti sperimentali, si può notare che la sensibilità (segnale/concentrazione) è circa uguale a 15 u.a. (stima grafica), ed è costante fino a livelli di concentrazione dell’ordine di 70 nL/L. Il range dinamico sembra estendersi da 0,1 nL/L a circa di 90 nL/L.
Il range lineare si estende, approssimativamente, fino a C = 70-80 nL/L.
10 0 10 20 30 40 50 60 70 80 90 100 110
0
500
1000
1500
Concentrazione (nl/l)
Segnale
(u.a
.)
.

67
CRITERI NUMERICI PER LA SCELTA DI UN METODO ANALITICO
Criterio Cifra di merito
PrecisioneDeviazione standard assoluta, deviazione standard relativa, coefficiente di variazione, varianza
Accuratezza Errore sistematico assoluto, errore sistematico relativo
Sensibilità Sensibilità di calibrazione, sensibilità analitica
Limite di rivelabilità Bianco più tre volte la deviazione standard
Intervallo di concentrazione
Dalla concentrazione relativa al limite di misurazione quantitativa (LOQ) alla concentrazione limite di risposta lineare (LOL)
Seletività Coefficiente di selettività

68
5.4.6 Stima dell'incertezza di misura
5.4.6.1 Un laboratorio di taratura, o un laboratorio di prova che esegue le
proprie tarature, deve avere e deve applicare una procedura per stimare
l'incertezza di misura per tutte le tarature e tipi di taratura.
5.4.6.2 I laboratori di prova devono avere e devono applicare procedure
per stimare l'incertezza delle misure. In certi casi la natura dei metodi di
prova può escludere il calcolo dell'incertezza di misura rigoroso e valido dal
punto di vista metrologico e statistico. In questi casi il laboratorio deve
almeno tentare di identificare tutte le componenti dell'incertezza e fare una
stima ragionevole, e deve garantire che l'espressione del risultato non
fornisca un'impressione errata dell'incertezza. Una stima ragionevole deve
essere basata sulla conoscenza del metodo e sullo scopo della misura e
deve far uso, per esempio, delle esperienze precedenti e della validazione
dei dati.
(dalla UNI CEI EN ISO/IEC 17025)
L’importanza dell’incertezza di misurazione è stata pienamente recepita
dalla norma UNI CEI EN ISO/IEC 17025.

69
Nota 1
Il livello di rigore necessario in una stima dell'incertezza di misura dipende da
fattori come:
- i requisiti dei metodo di prova;
- i requisiti dei cliente;
- l'esistenza di limiti stretti su cui sono basate le decisioni della
conformità ad una specifica.
Nota 2
In quei casi in cui un metodo di prova ben conosciuto specifica i limiti delle
maggiori sorgenti di incertezza e specifica la forma di presentazione dei
risultati calcolati, si ritiene che il laboratorio abbia soddisfatto questo punto,
seguendo i metodi di prova e le istruzioni per la presentazione dei risultati
(vedere 5.10).
(dalla UNI CEI EN ISO/IEC 17025)

70
5.4.6.3 Quando si stima l'incertezza di misura, devono essere prese in
considerazione, utilizzando appropriati metodi di analisi, tutte le
componenti dell'incertezza che sono di rilievo in una data situazione.
Nota 1
Le fonti che contribuiscono all'incertezza di misura includono, in modo non
esaustivo, i campioni di riferimento e i materiali di riferimento utilizzati, i
metodi e le apparecchiature utilizzate, le condizioni ambientali e le condizioni degli oggetti da provare o da tarare, e l'operatore.
Nota 2
Il comportamento previsto a lungo termine dell'oggetto sottoposto a prova
e/o taratura non è, di regola, preso in considerazione quando si stima
l'incertezza di misura.
Nota 3
Per ulteriori informazioni vedere ISO 5725 e la Guida all'espressione
dell'incertezza di misura (vedere bibliografia).
(dalla UNI CEI EN ISO/IEC 17025)

71
5.10.3 Rapporti di prova
5.10.3.1 In aggiunta a quanto indicato in 5.10.2, i rapporti di prova
devono includere, se necessario per l'interpretazione dei risultati, quanto
segue:
…
c) quando applicabile, una dichiarazione circa l'incertezza di misura
stimata; informazioni circa l'incertezza sono necessarie nel rapporto di
prova quando ciò influisce sulla validità o sull'applicazione dei risultati di
prova, quando le istruzioni dei cliente lo richiedono, o quando
l'incertezza ha influenza sulla conformità con un limite specificato;
…
(dalla UNI CEI EN ISO/IEC 17025)

72
Un primo modello, proposto da Wernimont [1], prevedeva la valutazione dell’UOM per mezzo delle stime di precisione eseguite in prove di confronto interlaboratorio (methodmethod--performance interperformance inter--
laboratory studieslaboratory studies).).
1. G.T. Wernimont, Use of statistics to develop and evaluate analytical methods, AOAC, Arlington, VA, (1985)
2. ISO, Guide to the expression of uncertainty in measurement, Geneva (1993)
3. EURACHEM, Quantifying uncertainty in analytical measurement, 1st Ed. (1995)
1. G.T. Wernimont, Use of statistics to develop and evaluate analytical methods, AOAC, Arlington, VA, (1985)
2. ISO, Guide to the expression of uncertainty in measurement, Geneva (1993)
3. EURACHEM, Quantifying uncertainty in analytical measurement, 1st Ed. (1995)
Dai primi anni ‘80 sono stati proposti numerosi modelli per la stima dell’incertezza di misurazione.
Successivamente, ISO ha proposto un modello completamente differente, noto come bottombottom--upup (o erroro error--budget, o budget, o componentcomponent--byby--
componentcomponent ), basato sui principi di propagazione degli errori. Le linee guida del modello sono descritte nella Guide to the expression of uncertainty in measurement [2], (nota come GUM).
Il modello bottom-up è stato poi adottato da EURACHEM [3].

73
Due anni dopo NMKL (Nordisk Metodik Komité for Levnedsmidler), giudicando il modello bottom-up più adatto a misurazioni fisiche che a misurazioni chimiche, ha sviluppato un modello alternativo piùsemplice e, allo stesso tempo, utile alla stima dell’incertezza complessiva connessa con l’intera procedura analitica totale [5].
Almeno in linea di principio, la norma UNI CEI EN ISO/IEC 17025 ha adottato il modello bottom-up, riferendosi esplicitamente alla GUM nelle sue linee guida.
4. Analytical Methods Committee, Uncertainty of Measurement: Implications of Its Use in Analytical Science, Analyst, 129 (1995) 2303
5. NMKL Procedure N. 5, Estimation and expression of measurement uncertainty in chemical analysis,
NMKL (1997)
4. Analytical Methods Committee, Uncertainty of Measurement: Implications of Its Use in Analytical Science, Analyst, 129 (1995) 2303
5. NMKL Procedure N. 5, Estimation and expression of measurement uncertainty in chemical analysis,
NMKL (1997)
In seguito alle perplessità avanzate da numerosi operatori, l’Analytical Methods Committee (AMC) della Royal Society of Chemistry (RSC) ha proposto un modello [4], noto come top-down, basato su quello di Wernimont.

74
Infine, Barwick ed Ellison [8] hanno predisposto un protocollo per utilizzare i
risultati degli studi di validazione nella stima dell’incertezza di misura. In
pratica l’approccio, sempre del tipo bottom-up integrato, descrive come i
dati ottenuti nei test di robustezza permettano di valutare opportunamente
tutte le sorgenti d’incertezza non considerate dagli studi di esattezza e
precisione.
6. EURACHEM, Quantifying uncertainty in analytical measurement, 2nd Ed. (2000)
7. Report on the FAO, IAEA, AOAC Int., IUPAC International Workshop on Principles and Practices of Method Validation, Budapest (1999)
8. V.J. Barwick, S.R.L. Ellison, VAM Project 3.2.1 Development and Harmonisation of Measurement
Uncertainty Principles. Part d. Protocol for uncertainty evaluation from validation data. Version 5.1
(2000)
6. EURACHEM, Quantifying uncertainty in analytical measurement, 2nd Ed. (2000)
7. Report on the FAO, IAEA, AOAC Int., IUPAC International Workshop on Principles and Practices of Method Validation, Budapest (1999)
8. V.J. Barwick, S.R.L. Ellison, VAM Project 3.2.1 Development and Harmonisation of Measurement
Uncertainty Principles. Part d. Protocol for uncertainty evaluation from validation data. Version 5.1
(2000)
Successivamente, la seconda edizione della Guida EURACHEM [6] e la
IUPAC [7] hanno proposto, per la stima dell’incertezza di misurazione, di
usare anche i dati acquisiti nel corso di studi di validazione (modello
bottom-up integrato).

75
INCERTEZZA DI MISURAZIONE
La norma ISO 25, Guide to the Expression of Uncertainty in Measurement
(ISO, Geneva, 1993), definisce l’incertezza di misurazione (UOM)* come
un parametro, associato al risultato di una misurazione, che
caratterizza la dispersione dei valori che possono essere
ragionevolmente attribuiti al misurando qualora siano state
considerate tutte le sorgenti d’errore.
Il risultato di una misurazione rappresenta la migliore stima del valore del
misurando e l’incertezza, valutata considerando tutte le sorgenti d’errore,
quantifica la qualità del risultato.
Una misura non completata dalla sua incertezza non può essere
confrontata né con altre misure né con valori di riferimento o con limiti
legali o composizionali.
* Sebbene Uncertainty of measurement debba essere correttamente tradotto in Incertezza di misurazione, è frequente l’uso di Incertezza di misura.
* Sebbene Uncertainty of measurement debba essere correttamente tradotto in Incertezza di misurazione, è frequente l’uso di Incertezza di misura.

76
Procedura per la valutazione della ripetibilità:
• analizzare 10 (N) standard, o materiali di riferimento o bianchifortificati indipendenti a diversi livelli di concentrazione entro il range dinamico (stesso operatore, strumento, laboratorio; tempo limitato);
• determinare la deviazione standard e calcolare il limite di ripetibiltà:
r,12tr
2
σ⋅⋅=υ−α
dove t1-α/2,ν è la t di Student per il livello di fiducia desiderato e ν = (N-1) gradi di libertà. In pratica, si accetta come possibile l’uso di ν = ∞ e quindi, per 1-α = 0,95, si usa t1-α/2,∞ = 1,96 ≈ 2); σr è la deviazione standard della ripetibilità.

77
L’attuale coesistenza di diversi modelli per la valutazione dell’incertezza di misurazione, e l’aumento dei costi e tempi di analisi derivante dalla loro applicazione, hanno trasformato la stima dell’incertezza di misurazione in uno dei maggiori problemi affrontati dai laboratori che vogliono introdurre un sistema di controllo qualità, o che hanno come obiettivo l’accreditamento dei loro metodi di analisi.
L’attuale coesistenza di diversi modelli per la valutazione dell’incertezza di misurazione, e l’aumento dei costi e tempi di analisi derivante dalla loro applicazione, hanno trasformato la stima dell’incertezza di misurazione in uno dei maggiori problemi affrontati dai laboratori che vogliono introdurre un sistema di controllo qualità, o che hanno come obiettivo l’accreditamento dei loro metodi di analisi.

















