

Malattie Malattie monofattorialimonofattoriali nellnell’’uomouomo
http://www.ncbi.nlm.nih.gov/omim

Esempi di patologie a trasmissione Esempi di patologie a trasmissione autosomicaautosomica dominantedominante
�� AcondroplasiaAcondroplasia
�� Corea di Corea di HuntingtonHuntington
�� Neurofibromatosi Tipo1Neurofibromatosi Tipo1
� Nelle forme familiari, ogni figlio/a ha un genitore affetto.
� Spesso il paziente affetto ha una diminuita capacità di riprodursi. La malattia è quindi mantenuta nella popolazione per la comparsa di mutazioni de-novo (figli affetti con genitori sani).
� Gli omozigoti sono rarissimi se non in presenza di matrimonio tra individui affetti o per consanguineità. Hanno in genere una sintomatologia più severa rispetto all’eterozigote.
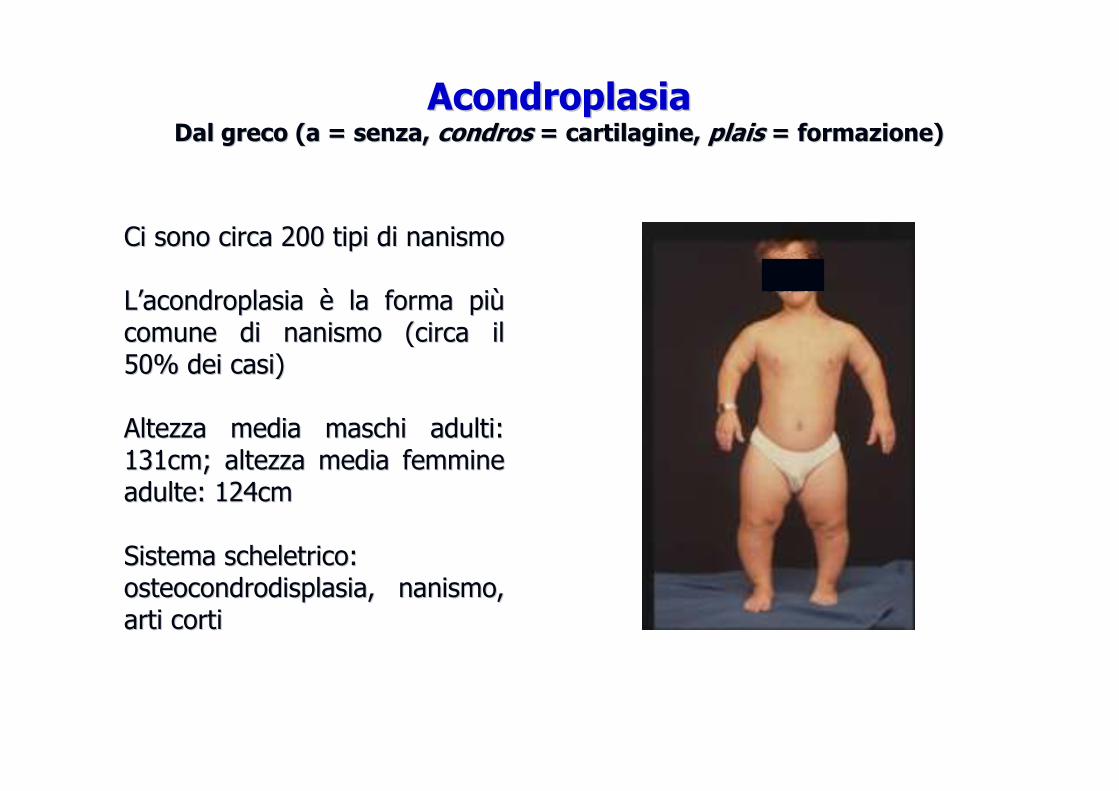
Ci sono circa 200 tipi di nanismoCi sono circa 200 tipi di nanismo
LL’’acondroplasia acondroplasia èè la forma pila forma piùùcomune di nanismo (circa il comune di nanismo (circa il 50% dei casi)50% dei casi)
Altezza media maschi adulti: Altezza media maschi adulti: 131cm; altezza media femmine 131cm; altezza media femmine adulte: 124cmadulte: 124cm
Sistema scheletrico:Sistema scheletrico:osteocondrodisplasiaosteocondrodisplasia, nanismo, , nanismo, arti cortiarti corti
AcondroplasiaAcondroplasiaDal greco (a = senza, Dal greco (a = senza, condroscondros = cartilagine, = cartilagine, plaisplais = formazione)= formazione)

�� Malattia genetica caratterizzata da un mancato sviluppo armonicoMalattia genetica caratterizzata da un mancato sviluppo armonico
della cartilagine di accrescimento delle ossa lunghe degli arti.della cartilagine di accrescimento delle ossa lunghe degli arti.
�� Letteralmente significa Letteralmente significa ““senza cartilaginesenza cartilagine”” anche se in realtanche se in realtàà il il
difetto difetto èè nella conversione della cartilagine in osso, specialmente nella conversione della cartilagine in osso, specialmente
nelle ossa lunghenelle ossa lunghe
�� Frequenza: 1/25.000 natiFrequenza: 1/25.000 nati
�� Causata da mutazioni del Causata da mutazioni del gene FGFR3gene FGFR3 (sul (sul cromosoma 4, cromosoma 4,
regione 4p16.3) che lo rendono regione 4p16.3) che lo rendono costitutivamentecostitutivamente attivo attivo �� allele allele
letale allo stato omozigoteletale allo stato omozigote

FGFR3 FGFR3 = = FibroblastFibroblast GrowthGrowth FactorFactor ReceptorReceptor 33
Recettore del fattore di crescita dei fibroblasti: proteina Recettore del fattore di crescita dei fibroblasti: proteina transmembranatransmembrana con attivitcon attivitàà
tirosintirosin chinasicachinasica e tre domini e tre domini IgIg--likelike
EE’’ un un regolatore negativo della proliferazione e del differenziamento regolatore negativo della proliferazione e del differenziamento dei dei condrociticondrociti
della cartilagine di accrescimentodella cartilagine di accrescimento; ; èè attivato dalle mutazioni presenti nei soggetti attivato dalle mutazioni presenti nei soggetti
con acondroplasia. Queste mutazioni sono da considerare del tipocon acondroplasia. Queste mutazioni sono da considerare del tipo con "guadagno con "guadagno
di funzione" (di funzione" (gaingain ofof functionfunction), perch), perchéé il recettore diventa piil recettore diventa piùù efficace nello efficace nello
svolgimento del suo ruolo inibitorio.svolgimento del suo ruolo inibitorio.
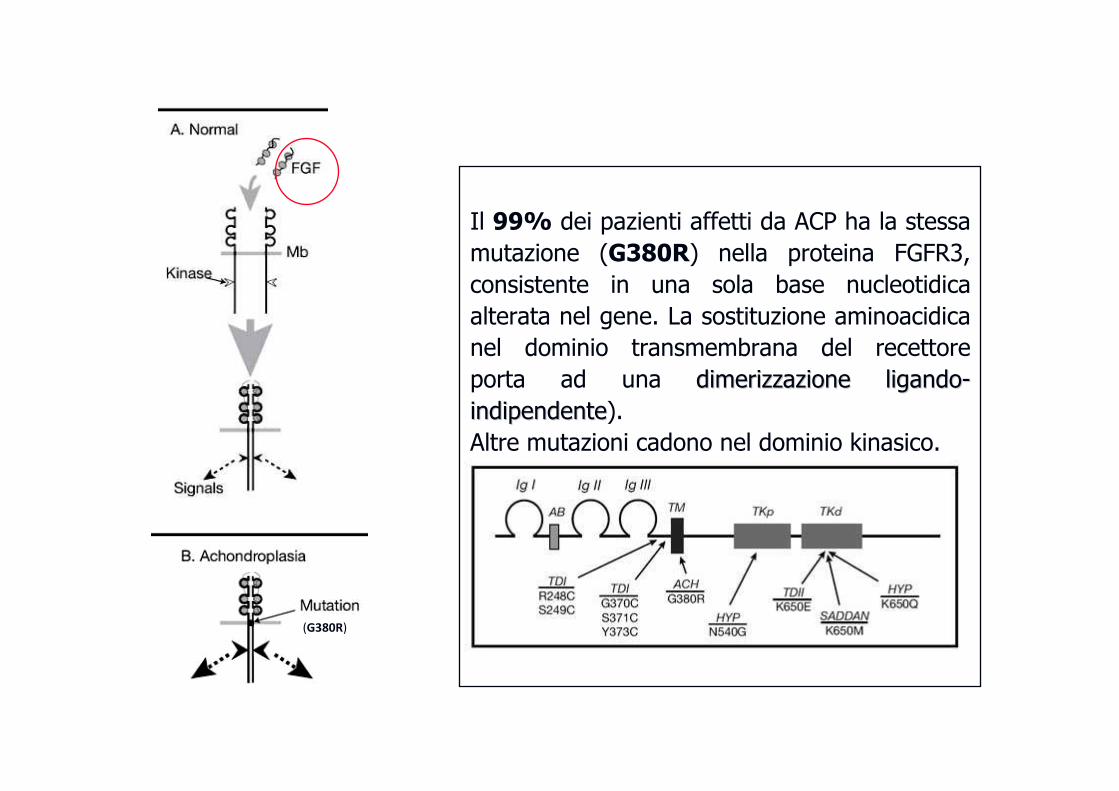
Il 99% dei pazienti affetti da ACP ha la stessa
mutazione (G380R) nella proteina FGFR3,
consistente in una sola base nucleotidica
alterata nel gene. La sostituzione aminoacidica
nel dominio transmembrana del recettore
porta ad una dimerizzazionedimerizzazione ligandoligando--
indipendenteindipendente).
Altre mutazioni cadono nel dominio kinasico.
(G380R)

EreditarietEreditarietàà
� EreditEreditàà aautosomicautosomica dominante a penetranza completadominante a penetranza completa
Una persona affetta da ACP, insieme ad una normale, hanno il 50% di probabilità di avere un figlio affetto.
Una coppia in cui entrambi i genitori sono affetti da ACP ha 1 probabilità su 4 di concepire un figlio/a
affetto da una grave forma di acondroplasia letale alla nascita (acondroplasia omozigote); 1 probabilità su
4 di avere figli normali e 1 probabilità su 2 di avere un figlio affetto
�� Effetto dellEffetto dell’’etetàà paterna per le mutazioni depaterna per le mutazioni de--novonovo
Circa 9 casi su 10 i pazienti affetti da ACP nascono da genitori assolutamente normali, a causa di
alterazioni nel gene che avvengono di solito negli spermatozoi (mutazioni mutazioni dede--novo)novo), correlati ad una avanzata età paterna.
DiagnosiDiagnosi
� Amplificazione per PCR del DNA genomico e Amplificazione per PCR del DNA genomico e sequenziamentosequenziamento degli degli esoni dove sono localizzate le mutazioni. esoni dove sono localizzate le mutazioni.
Alcune mutazioni danno origine a RFLP (es. G380R � la mutazione crea sito di restrizione per l’enzima SfcI)
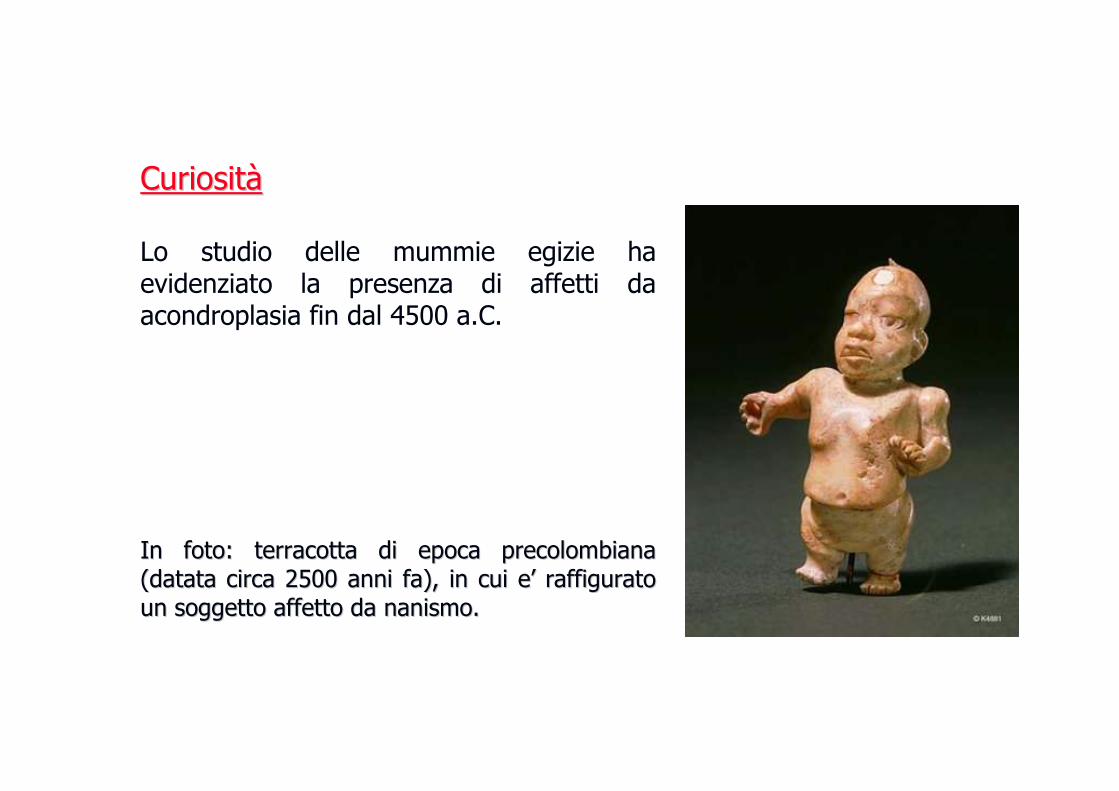
CuriositCuriositàà
Lo studio delle mummie egizie ha evidenziato la presenza di affetti da acondroplasia fin dal 4500 a.C.
In foto: terracotta di epoca precolombiana In foto: terracotta di epoca precolombiana (datata circa 2500 anni fa), in cui e(datata circa 2500 anni fa), in cui e’’ raffigurato raffigurato un soggetto affetto da nanismo.un soggetto affetto da nanismo.

Neurofibromatosi di Tipo1Neurofibromatosi di Tipo1
�� Malattia Malattia autosomicaautosomica dominante ad dominante ad espressivitespressivitàà variabile variabile (macchie caff(macchie caffèè--
latte/tumori fibromatosi della pelle/tumori maligni del SNC) latte/tumori fibromatosi della pelle/tumori maligni del SNC)
�� Frequenza: 1/3000 natiFrequenza: 1/3000 nati
�� Almeno il 50% degli affetti ha una storia familiare di neurofibrAlmeno il 50% degli affetti ha una storia familiare di neurofibromatosi 1 omatosi 1
documentata; nella restante parte dei casi si tratta di mutaziondocumentata; nella restante parte dei casi si tratta di mutazioni i dede--novonovo
�� Forme di Forme di mosaicismomosaicismo somatico (meno gravi)somatico (meno gravi)
�� Il gene della neurofibromatosi di tipo 1 (Il gene della neurofibromatosi di tipo 1 (NF1NF1) ) èè localizzato sul cromosoma localizzato sul cromosoma
1717 (17q11.2) e codifica per una proteina ((17q11.2) e codifica per una proteina (neurofibrominaneurofibromina) con attivit) con attivitàà inibitoria inibitoria
sulla crescita tumorale (inibisce l'sulla crescita tumorale (inibisce l'oncoproteinaoncoproteina p21ras), cosa che la fa p21ras), cosa che la fa
appartenere alla famiglia dei geni appartenere alla famiglia dei geni oncosoppressorioncosoppressori. La funzione della proteina si . La funzione della proteina si
riduce o viene alterata in seguito a mutazione.riduce o viene alterata in seguito a mutazione.
�� Le mutazioni piLe mutazioni piùù frequenti sono frequenti sono mutazioni puntiformimutazioni puntiformi e e ampie ampie delezionidelezioni
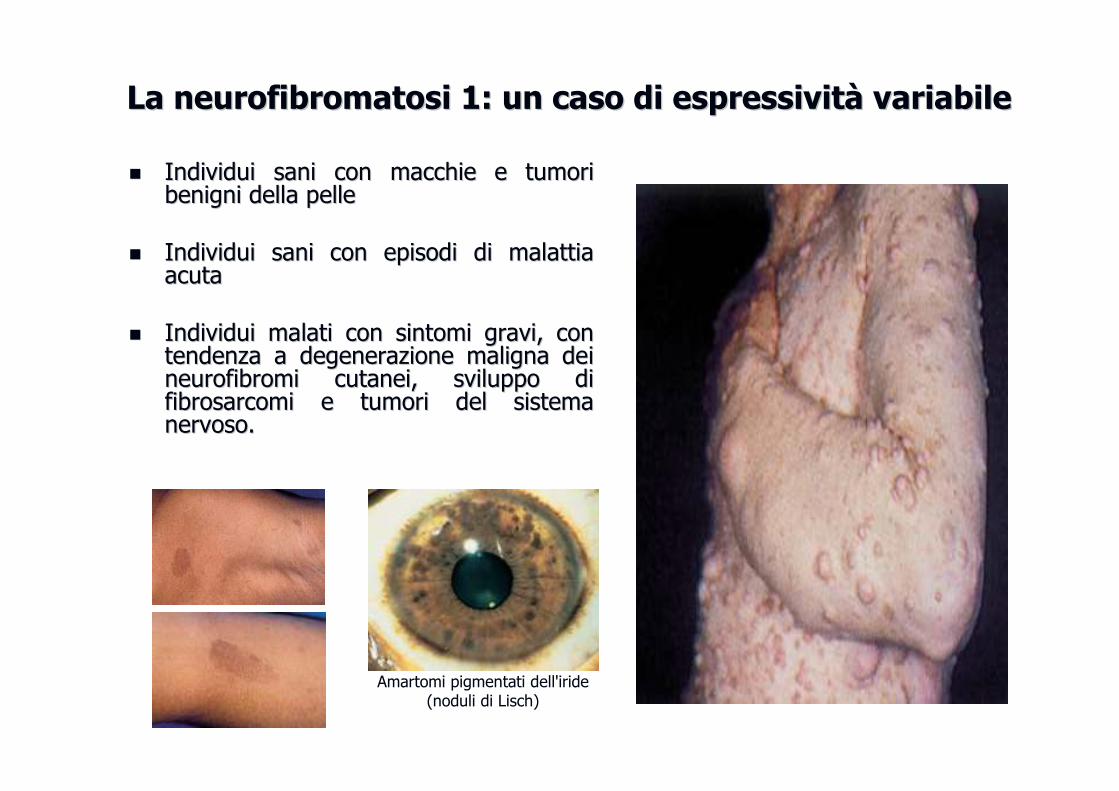
La neurofibromatosi 1: un caso di espressivitLa neurofibromatosi 1: un caso di espressivitàà variabilevariabile
�� IndividuiIndividui sanisani con con macchiemacchie e e tumoritumoribenignibenigni delladella pellepelle
�� IndividuiIndividui sanisani con con episodiepisodi didi malattiamalattiaacutaacuta
�� IndividuiIndividui malatimalati con con sintomisintomi gravigravi, con , con tendenzatendenza a a degenerazionedegenerazione malignamaligna deideineurofibromineurofibromi cutaneicutanei, , svilupposviluppo didifibrosarcomifibrosarcomi e e tumoritumori del del sistemasistemanervosonervoso. .
Amartomi pigmentati dell'iride (noduli di Lisch)

Esempi di patologie a trasmissione Esempi di patologie a trasmissione autosomicaautosomica recessivarecessiva
�� AlbinismoAlbinismo�� SorditSorditàà
�� Talassemia Talassemia �� FalcemiaFalcemia�� FenilchetonuriaFenilchetonuria�� Fibrosi cisticaFibrosi cistica
� I genitori sono solitamente eterozigoti (portatori sani), con il 25% di rischio di generare un figlio omozigote.
� Classicamente il pedigree mostra solo un membro della famiglia affetto o, al massimo, fratelli affetti.
� Per alcune patologie, la percentuale di portatori sani nella popolazione èestremamente elevata, spesso perché la condizione di eterozigoti dà un vantaggio selettivo (es. beta-talassemia), detto appunto vantaggio dell’eterozigote

Talassemia Talassemia (o microcitemia o anemia mediterranea)(o microcitemia o anemia mediterranea)
�� Disordini nella sintesi dellDisordini nella sintesi dell’’emoglobina dovuti allemoglobina dovuti all’’assenza o alla ridotta produzione di assenza o alla ridotta produzione di
catene di tipo catene di tipo αα o o ββ, che costituiscono l, che costituiscono l’’emoglobina umana adultaemoglobina umana adulta
�� Dal punto di vista clinico la talassemiaDal punto di vista clinico la talassemia èè contraddistinta da fragilitcontraddistinta da fragilitàà degli eritrociti, degli eritrociti,
microcitosimicrocitosi, anemia. , anemia.
�� ricambio troppo veloce dei globuli rossi che vengono distrutti mricambio troppo veloce dei globuli rossi che vengono distrutti molto rapidamente olto rapidamente
dalla milza (dalla milza (�� splenomegalia). splenomegalia).
�� lle periodiche trasfusioni di sangue e eritropoiesi inefficace proe periodiche trasfusioni di sangue e eritropoiesi inefficace provocano un vocano un
dannosissimo accumulo di ferro nei tessuti che va eliminato attrdannosissimo accumulo di ferro nei tessuti che va eliminato attraverso una terapia averso una terapia
ferroferro--chelantechelante
�� Frequenza: iFrequenza: in Italia n Italia èè molto rara, anche se molto rara, anche se èè di frequente riscontro nelle regioni del di frequente riscontro nelle regioni del
sud come Sicilia e Sardegna. In Sardegna il 12% degli individui sud come Sicilia e Sardegna. In Sardegna il 12% degli individui èè portatore sano di portatore sano di ββ--
talassemia. La malattia talassemia. La malattia èè frequente (3% nella popolazione mondiale) nell'Africa frequente (3% nella popolazione mondiale) nell'Africa
centrale, nel Sudcentrale, nel Sud--est asiatico, in Medio Oriente e in alcune zone dell'India.est asiatico, in Medio Oriente e in alcune zone dell'India.
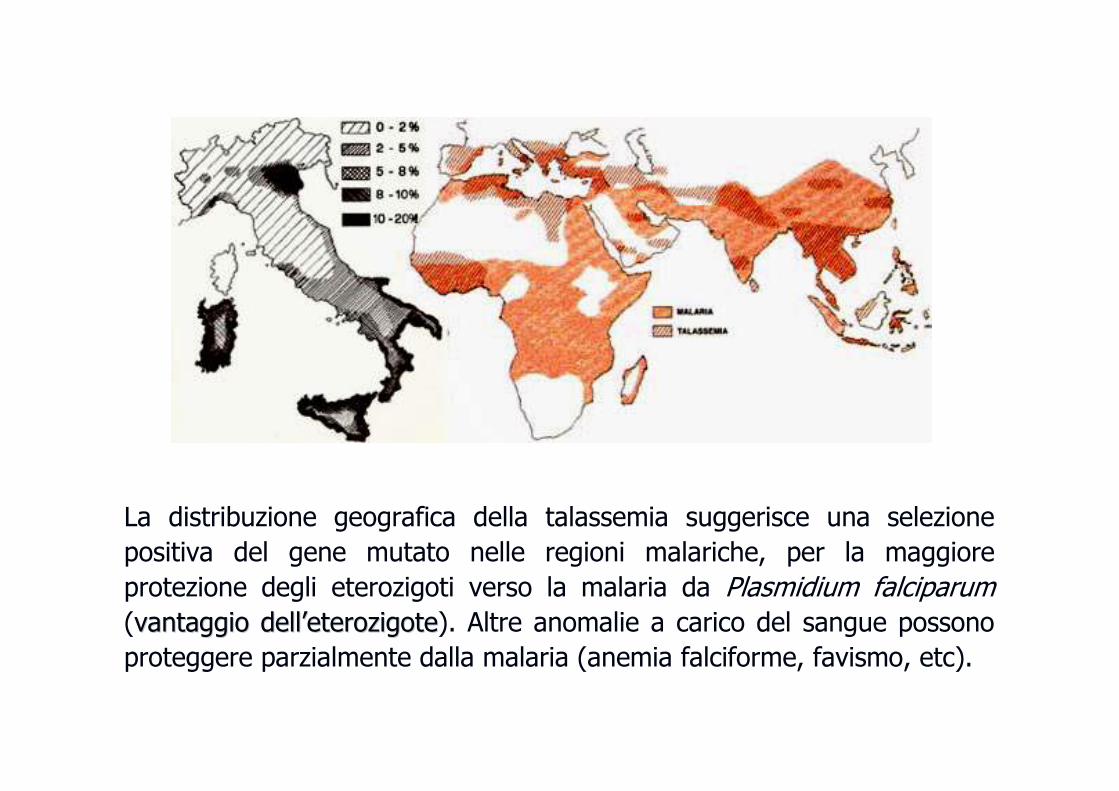
La distribuzione geografica della talassemia suggerisce una selezione
positiva del gene mutato nelle regioni malariche, per la maggiore
protezione degli eterozigoti verso la malaria da Plasmidium falciparum(vantaggio dellvantaggio dell’’eterozigoteeterozigote). Altre anomalie a carico del sangue possono
proteggere parzialmente dalla malaria (anemia falciforme, favismo, etc).
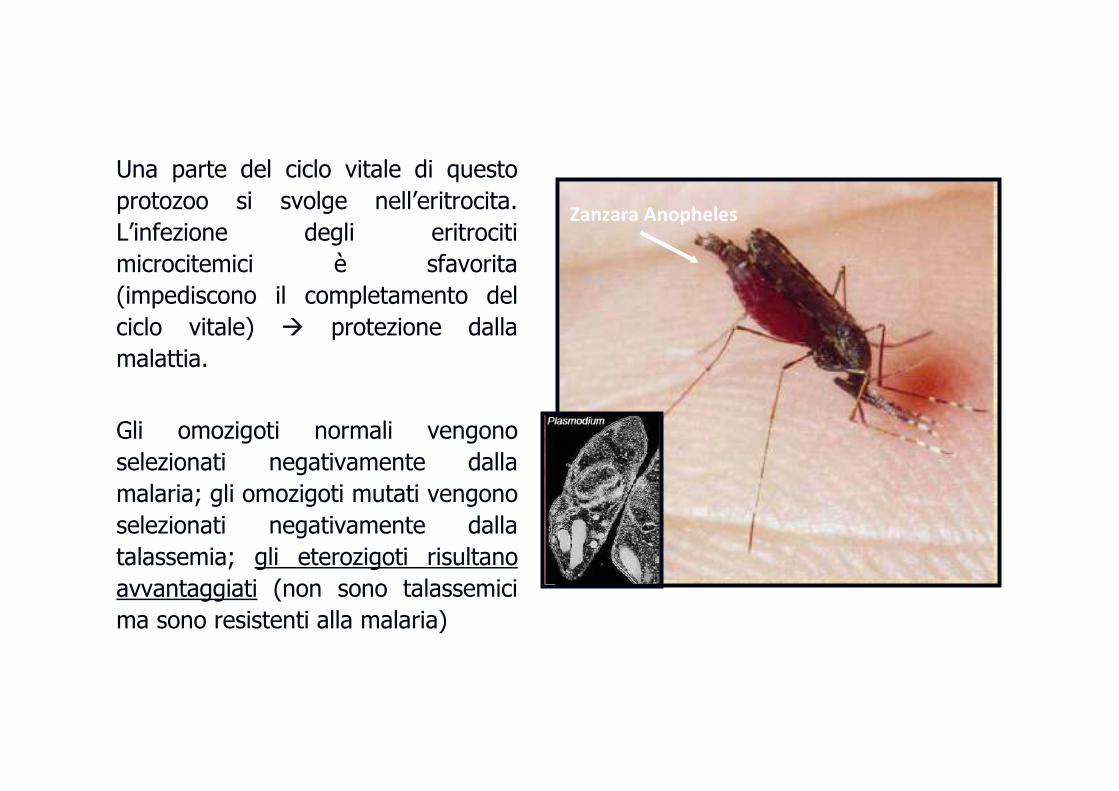
Una parte del ciclo vitale di questo
protozoo si svolge nell’eritrocita.
L’infezione degli eritrociti
microcitemici è sfavorita
(impediscono il completamento del
ciclo vitale) � protezione dalla
malattia.
Gli omozigoti normali vengono
selezionati negativamente dalla
malaria; gli omozigoti mutati vengono
selezionati negativamente dalla
talassemia; gli eterozigoti risultano
avvantaggiati (non sono talassemici
ma sono resistenti alla malaria)
Zanzara Anopheles
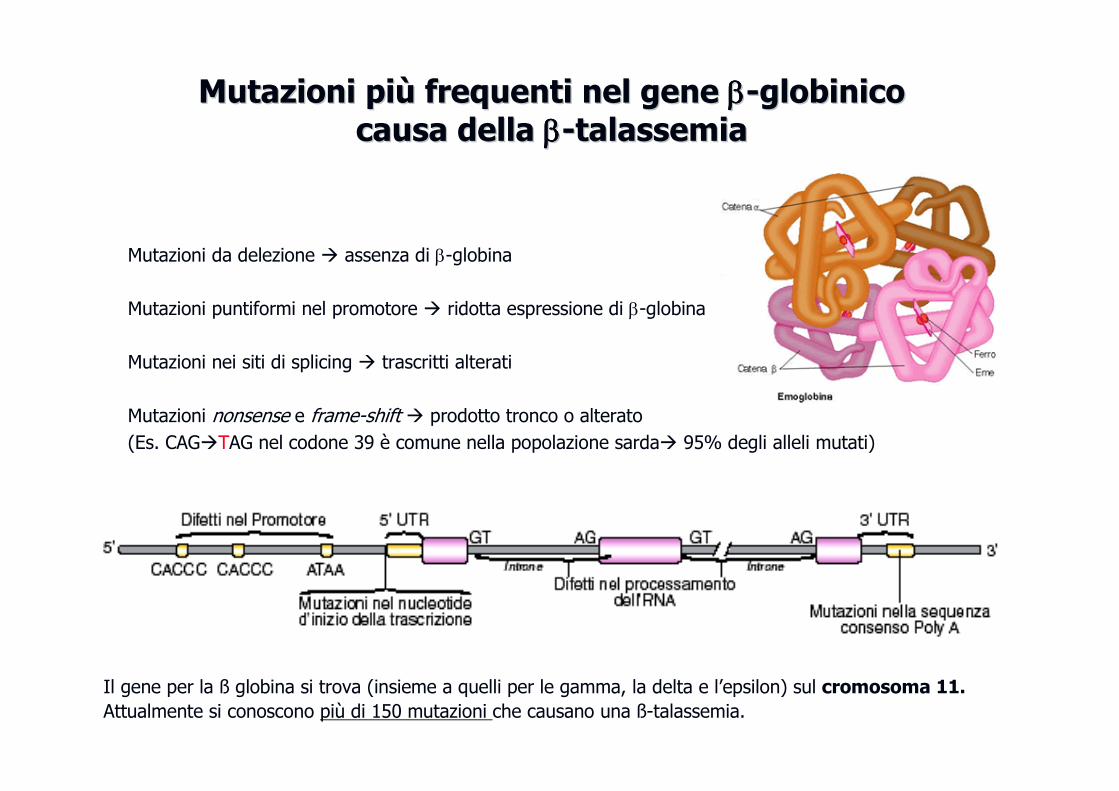
Mutazioni da delezione � assenza di β-globina
Mutazioni puntiformi nel promotore � ridotta espressione di β-globina
Mutazioni nei siti di splicing � trascritti alterati
Mutazioni nonsense e frame-shift � prodotto tronco o alterato
(Es. CAG�TAG nel codone 39 è comune nella popolazione sarda� 95% degli alleli mutati)
Mutazioni piMutazioni piùù frequenti nel gene frequenti nel gene ββββββββ--globinicoglobinicocausa della causa della ββββββββ--talassemiatalassemia
Il gene per la ß globina si trova (insieme a quelli per le gamma, la delta e l’epsilon) sul cromosoma 11.
Attualmente si conoscono più di 150 mutazioni che causano una ß-talassemia.

3 quadri clinici3 quadri clinici che si distinguono tra loro per gravità crescente:
� Portatore asintomatico (microcitemico): è caratterizzato da assenza di
sintomi clinici, aumento del numero dei globuli rossi con riduzione del loro
volume (da ciò il nome di microcitemia), riduzione della concentrazione di
emoglobina contenuta nei globuli rossi e alterazione della loro forma. Genotipo: eterozigote
� Talassemia intermedia: in questo gruppo eterogeneo di pazienti sono
compresi casi con gravità differente, da forme minime con lievi manifestazioni
cliniche a casi più gravi a volte simili alla forma grave. In questi casi c'è un
marcato aumento dell'emoglobina fetale. Genotipo: omozigote per un difetto lieve o
eterozigote per un difetto grave ed uno lieve.
� Talassemia major o malattia di Cooley: è la forma più grave di
microcitemia, caratterizzata da anemia marcata. Genotipo: omozigote per un difetto
grave o eter.ozigosi mista per due difetti gravi.
La terapia è diversa a seconda della gravità della malattia
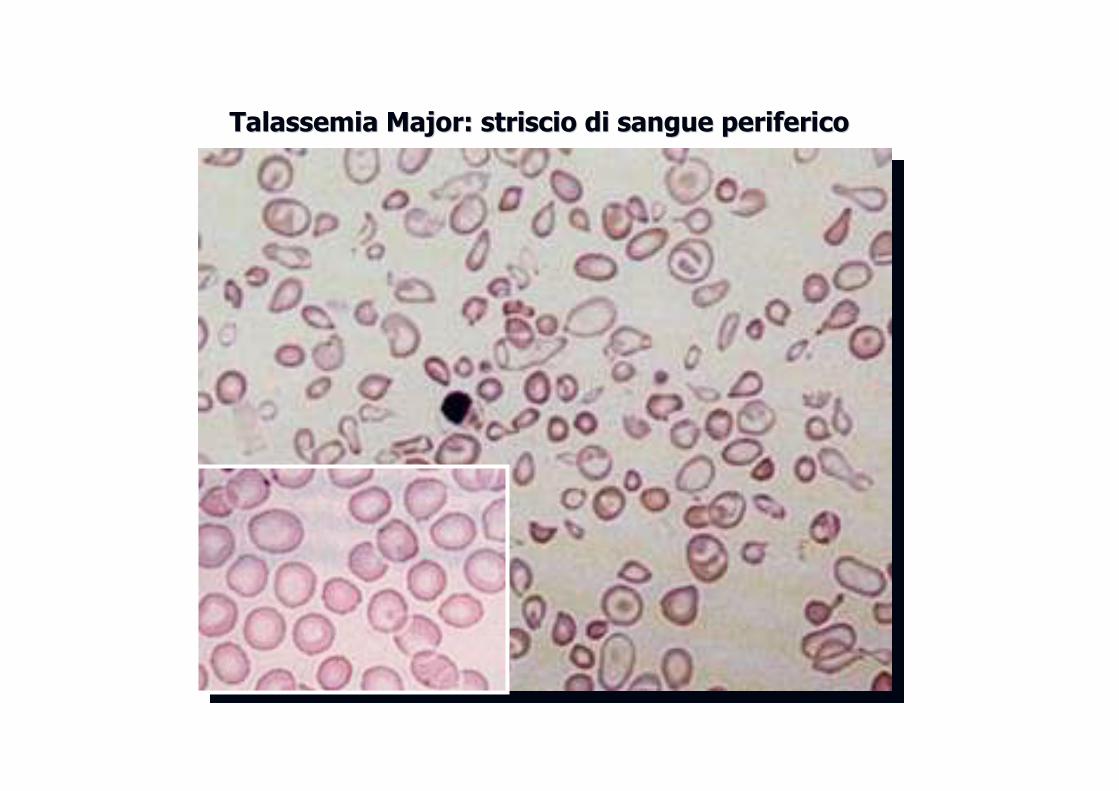
Talassemia Major: striscio di sangue perifericoTalassemia Major: striscio di sangue periferico

Anemia falciformeAnemia falciforme
�� Dovuta alla singola mutazione nel gene per la Dovuta alla singola mutazione nel gene per la
subunitsubunitàà ββ deldelll’’emoglobina emoglobina �� forma anomala, detta forma anomala, detta
emoglobina S (Gemoglobina S (GAAGG�� GGTTG nel codone 6= G nel codone 6= GluGlu��ValVal) )
�� Il quadro ematologico tipico, caratterizzato dalla Il quadro ematologico tipico, caratterizzato dalla presenza di globuli rossi a forma presenza di globuli rossi a forma
di falcedi falce, , èè dovuto alla polimerizzazione dell'emoglobina S con formazione ddovuto alla polimerizzazione dell'emoglobina S con formazione di fibre i fibre
all'interno del globulo rosso. Ne risulta 1) una all'interno del globulo rosso. Ne risulta 1) una rigiditrigiditàà dei globuli rossidei globuli rossi i quali tendono i quali tendono
ad occludere i vasi periferici e 2) una ad occludere i vasi periferici e 2) una ridotta vita media dei globuli rossi stessiridotta vita media dei globuli rossi stessi che che èè
alla base dell'anemia cronica.alla base dell'anemia cronica.
�� Dal punto di vista clinico la malattia presenta sintomi legati aDal punto di vista clinico la malattia presenta sintomi legati all'll'emolisi emolisi (distruzione) dei (distruzione) dei
globuli rossi e alle crisi da globuli rossi e alle crisi da occlusione dei vasi sanguigni. occlusione dei vasi sanguigni.
�� Frequente nelle regioni del Mediterraneo (soprattutto in Africa)Frequente nelle regioni del Mediterraneo (soprattutto in Africa)
�� AplosufficienzaAplosufficienza/insufficienza negli eterozigoti/insufficienza negli eterozigoti
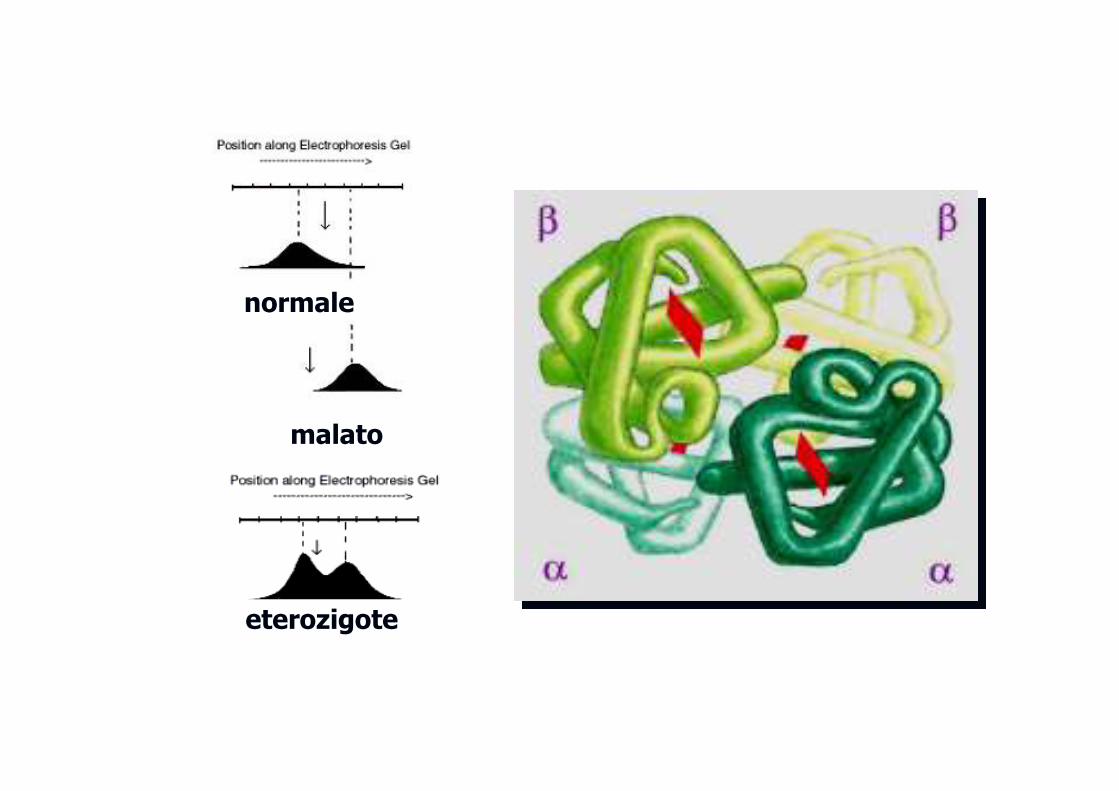
normale
malato
eterozigote

� E’ causata da mutazioni nel gene che codifica per l’enzima fenilalaninaidrossilasi (sul cromosoma 12cromosoma 12, 12q24.1). Frequenza: 1/10000
� Sono note 462 mutazioni del gene (soprattutto missense, nonsense ma anche delezioni, inserzioni, splicing mutations e mutazioni de novo)
Fenilchetonuria (PKU)Fenilchetonuria (PKU)
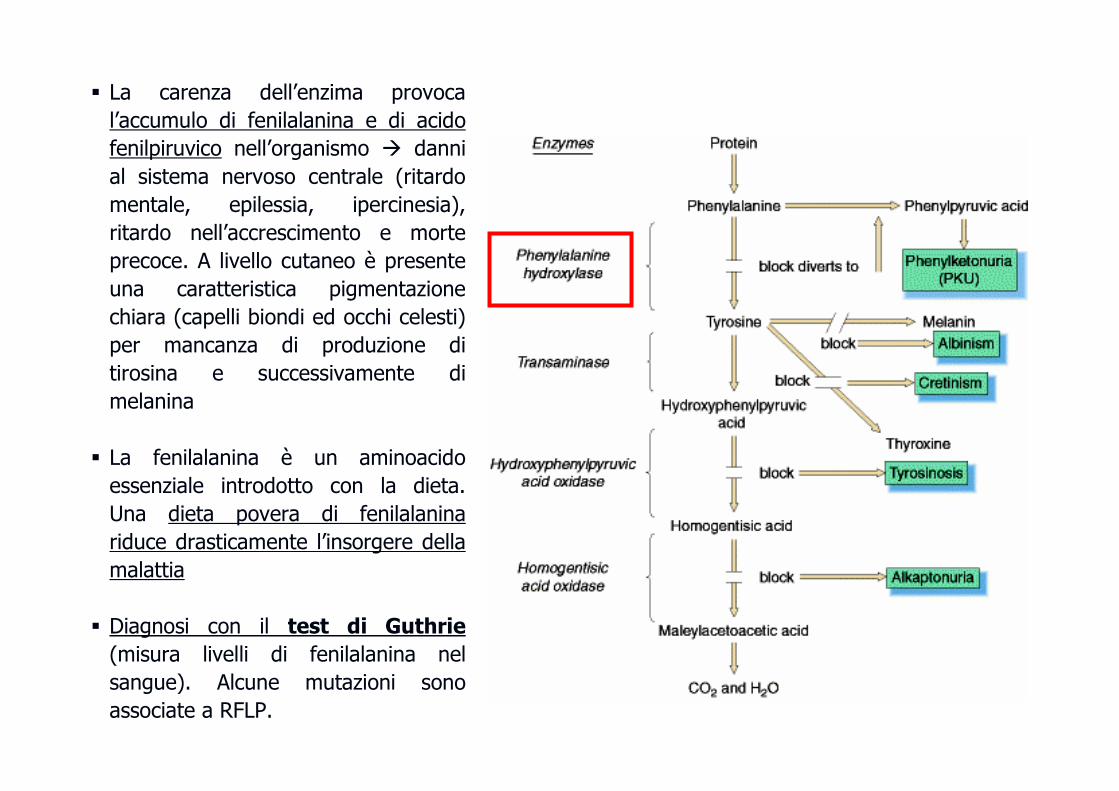
� La carenza dell’enzima provoca
l’accumulo di fenilalanina e di acido
fenilpiruvico nell’organismo � danni
al sistema nervoso centrale (ritardo
mentale, epilessia, ipercinesia),
ritardo nell’accrescimento e morte
precoce. A livello cutaneo è presente
una caratteristica pigmentazione
chiara (capelli biondi ed occhi celesti)
per mancanza di produzione di
tirosina e successivamente di
melanina
� La fenilalanina è un aminoacido
essenziale introdotto con la dieta.
Una dieta povera di fenilalanina
riduce drasticamente l’insorgere della
malattia
� Diagnosi con il test di Guthrie
(misura livelli di fenilalanina nel
sangue). Alcune mutazioni sono
associate a RFLP.

� E’ la più comune malattia autosomicaautosomica recessivarecessiva (1/2000 nati � ~5% degli individui sono portatori sani)
� Interessa numerosi organi, come polmoni, fegato, pancreas, intestino ed apparato riproduttivo
� Mostra espressivitespressivitàà variabilevariabile ed eterogeneiteterogeneitàà clinicaclinica
� Causata da mutazioni nel gene che codifica per la proteina CFTRproteina CFTR, localizzato sul cromosoma 7 cromosoma 7 (7q31.2).
Fibrosi cisticaFibrosi cistica
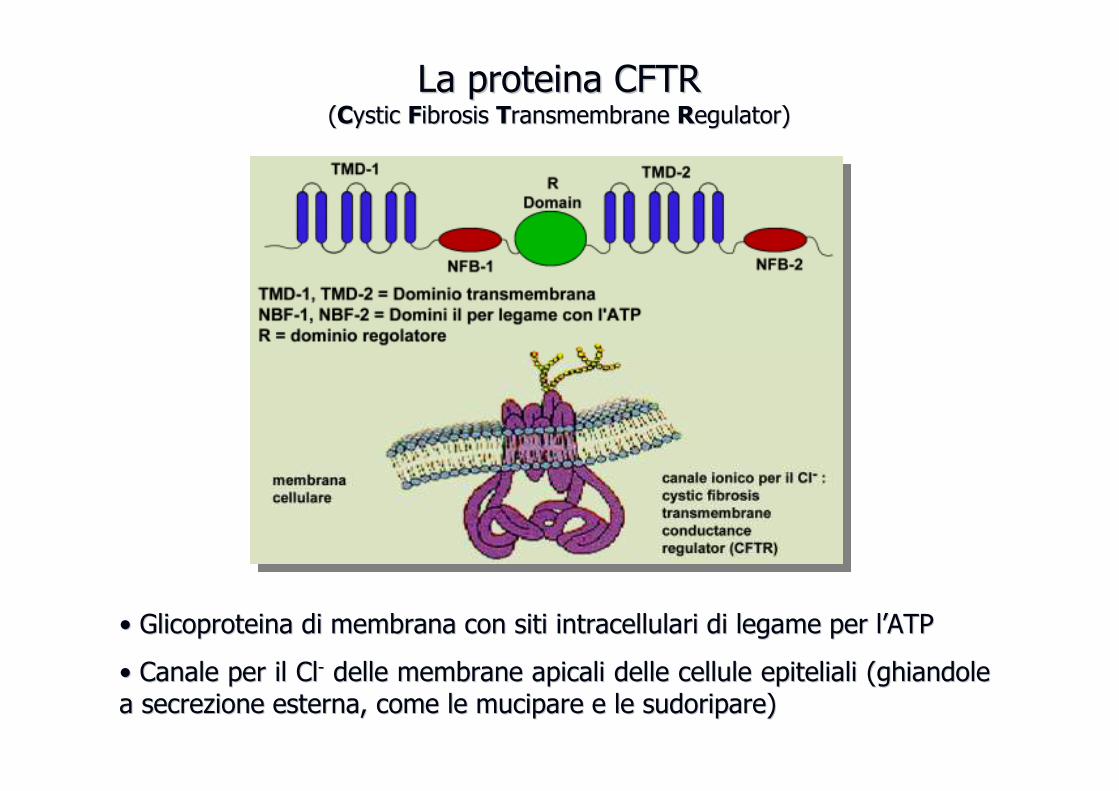
La proteina CFTRLa proteina CFTR((CCysticystic FFibrosisibrosis TTransmembraneransmembrane RRegulatoregulator))
•• GlicoproteinaGlicoproteina di membrana con siti intracellulari di legame per ldi membrana con siti intracellulari di legame per l’’ATP ATP
•• Canale per il ClCanale per il Cl-- delle membrane apicali delle cellule epiteliali (ghiandole delle membrane apicali delle cellule epiteliali (ghiandole a secrezione esterna, come le mucipare e le sudoripare)a secrezione esterna, come le mucipare e le sudoripare)
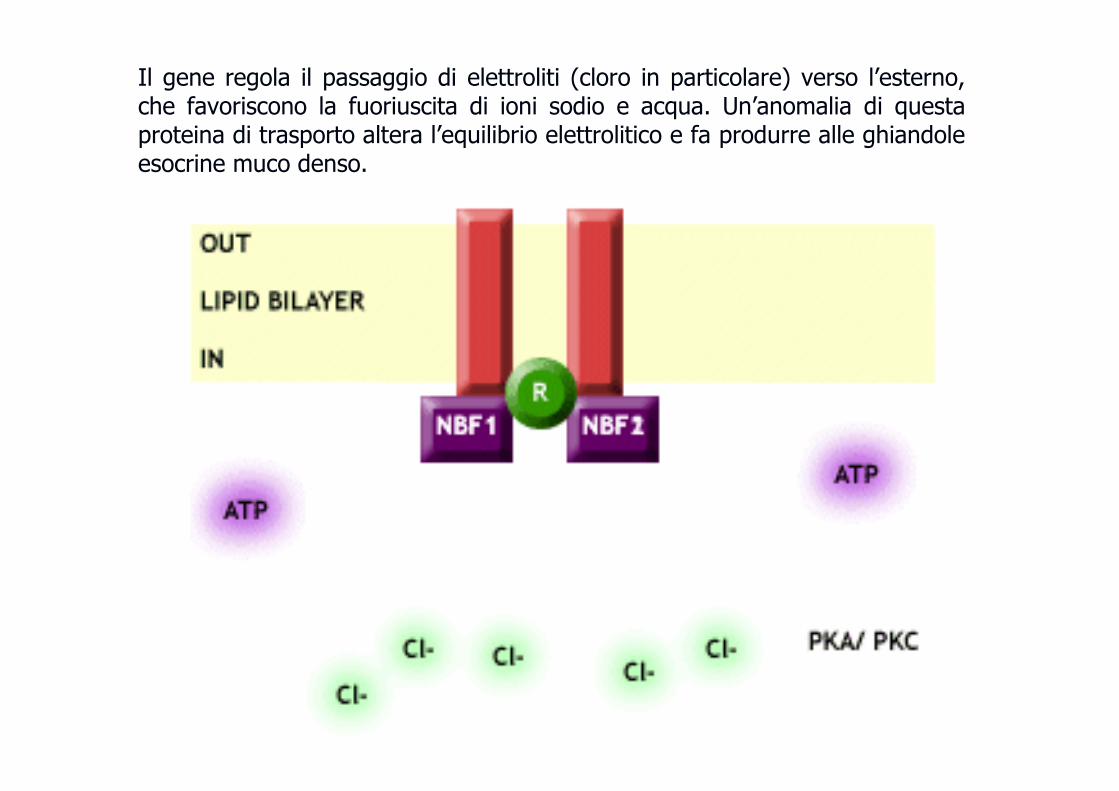
Il gene regola il passaggio di elettroliti (cloro in particolare) verso l’esterno, che favoriscono la fuoriuscita di ioni sodio e acqua. Un’anomalia di questa proteina di trasporto altera l’equilibrio elettrolitico e fa produrre alle ghiandole esocrine muco denso.
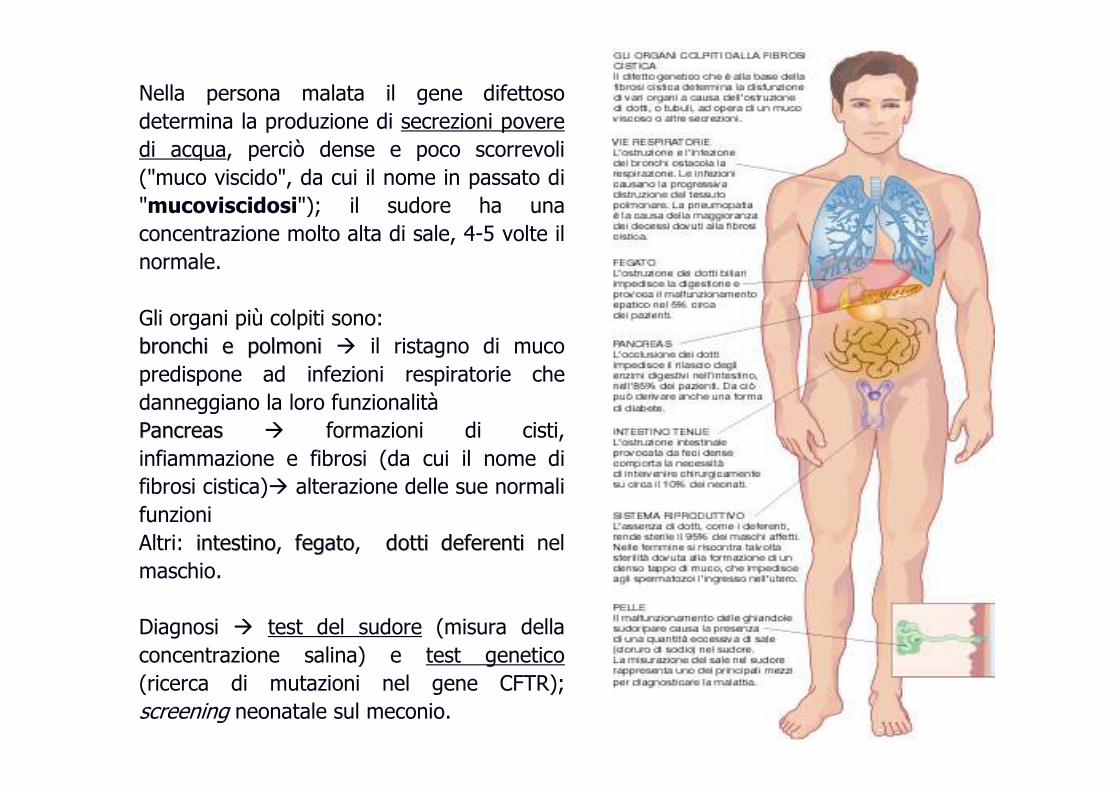
Nella persona malata il gene difettoso
determina la produzione di secrezioni povere
di acqua, perciò dense e poco scorrevoli
("muco viscido", da cui il nome in passato di
"mucoviscidosi"); il sudore ha una
concentrazione molto alta di sale, 4-5 volte il
normale.
Gli organi più colpiti sono:
bronchi e polmonibronchi e polmoni � il ristagno di muco
predispone ad infezioni respiratorie che
danneggiano la loro funzionalità
PancreasPancreas � formazioni di cisti,
infiammazione e fibrosi (da cui il nome di
fibrosi cistica)� alterazione delle sue normali
funzioni
Altri: intestinointestino, fegatofegato, dotti deferentidotti deferenti nel
maschio.
Diagnosi � test del sudore (misura della
concentrazione salina) e test genetico
(ricerca di mutazioni nel gene CFTR);
screening neonatale sul meconio.

Mutazioni del gene CFTRMutazioni del gene CFTR
Si presentano in modalità diversa nelle varie popolazioni, sia per tipologia che
per frequenza. Più di 1800 mutazioni note.
Alcune mutazioni fanno sì che essa non venga prodotta affatto, altre permettono
che venga prodotta una proteina poco funzionante o ridotta in quantità. Non di
tutte si conosce l'effetto clinico.
Queste mutazioni determinano una disfunzione della proteina CFTR attraverso
numerosi meccanismi molecolari, in base ai quali è possibile raggrupparle in 5
differenti classi
la mutazione più frequente in Italia

AAT ATC ATC TTT GGT GTT TCCAsn Ile Ile PhePhe Gly Val Ser
La mutazione La mutazione ∆∆F508F508
Consiste nella perdita di 3 basi con conseguente mancanza di unafenilalanina nella catena aminoacidica della proteina)
AAT ATC ATT GGT GTT TCCAsn Ile Ile Gly Val Ser
Wild-type
mutato
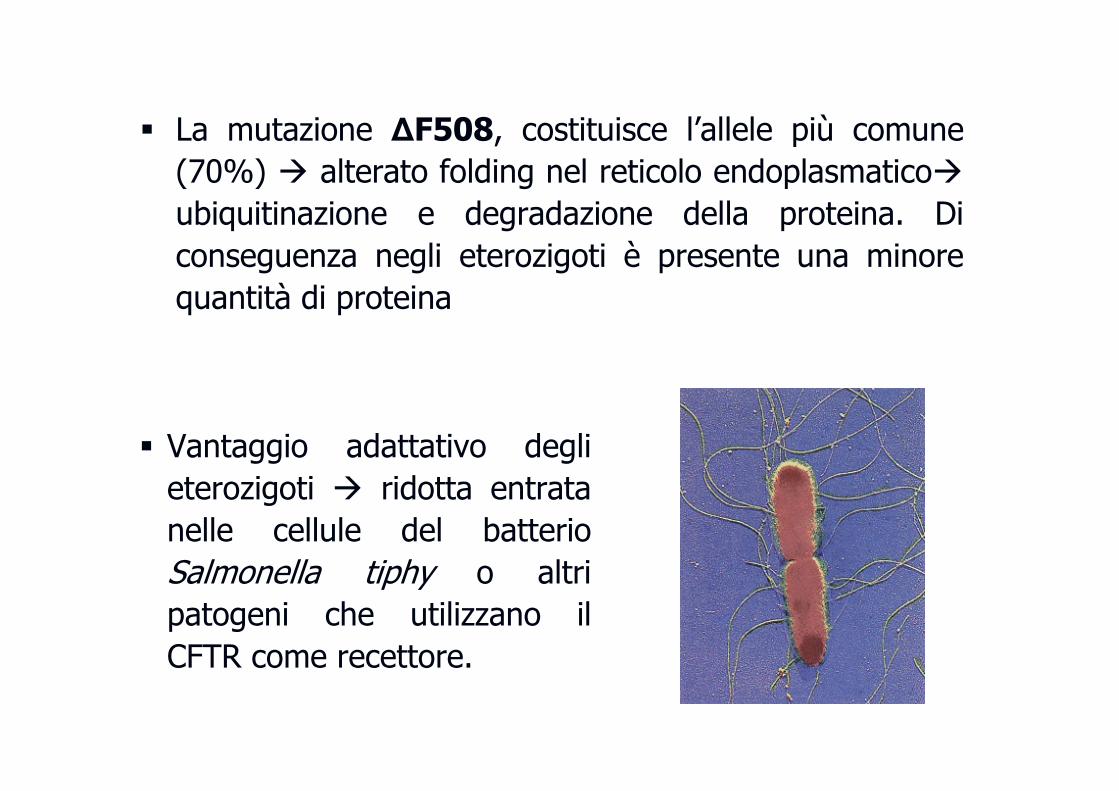
� La mutazione ∆F508, costituisce l’allele più comune
(70%) � alterato folding nel reticolo endoplasmatico�
ubiquitinazione e degradazione della proteina. Di
conseguenza negli eterozigoti è presente una minore
quantità di proteina
� Vantaggio adattativo degli
eterozigoti � ridotta entrata
nelle cellule del batterio
Salmonella tiphy o altri
patogeni che utilizzano il
CFTR come recettore.
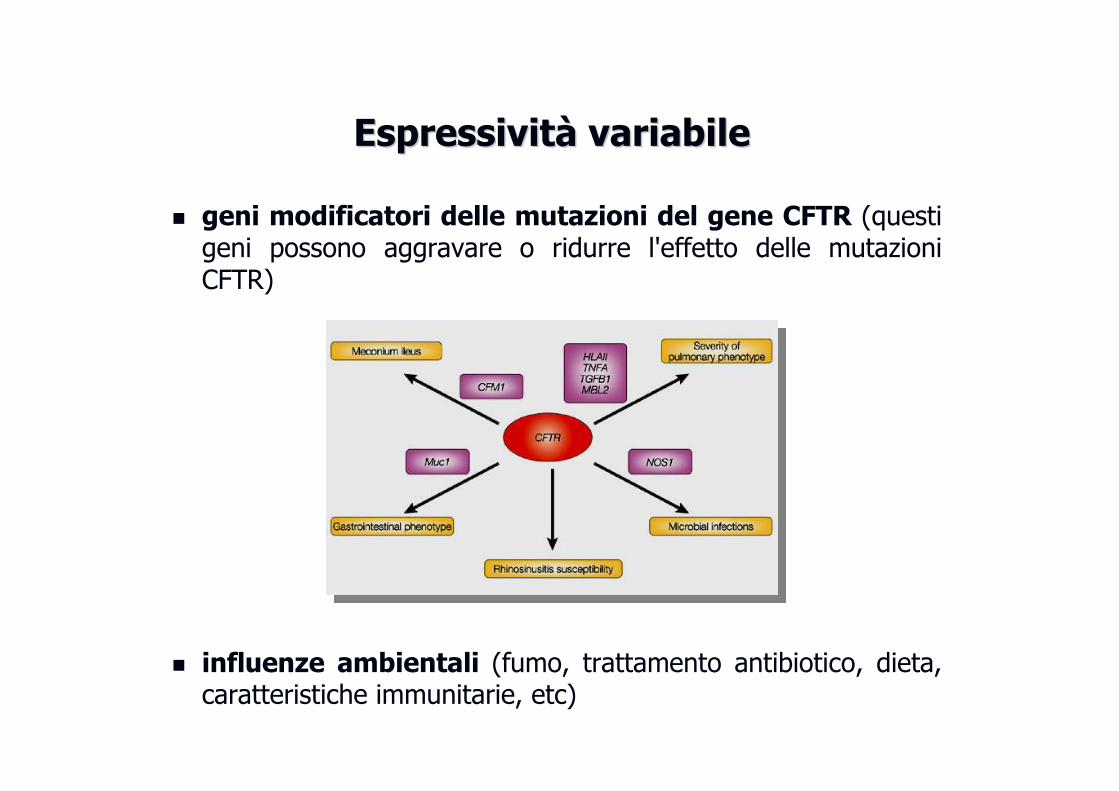
EspressivitEspressivitàà variabilevariabile
� geni modificatori delle mutazioni del gene CFTR (questi geni possono aggravare o ridurre l'effetto delle mutazioni CFTR)
� influenze ambientali (fumo, trattamento antibiotico, dieta, caratteristiche immunitarie, etc)

Esempi di patologie a trasmissione Esempi di patologie a trasmissione XX--linkedlinked recessivarecessiva
�� DaltonismoDaltonismo
�� Emofilia AEmofilia A
�� Distrofia di Distrofia di DuchenneDuchenne
�� FavismoFavismo
� Generalmente ristrette ai maschi, perché le femmine per manifestare la malattia devono essere omozigoti

Emofilia Emofilia
� Malattia con difetti più o meno gravi nella coagulazione del sangue
trasmessa come carattere legato al cromosoma X
� Emofilia A e B sono distinte dal punto di vista molecolare e
biochimico ma simili dal punto di vista clinico � anche minimi traumi
possono provocare emorragie gravi

Emofilia AEmofilia A
� E’ un difetto della coagulazione del sangue trasmesso come carattere legato al cromosoma X e dovuto ad un deficit del fattore VIII della coagulazione (gene F8gene F8, Xq28)
� La terapia si basa sulla somministrazione di concentrati di fattore VIII preparati da plasma umano o mediante la tecnica del DNA ricombinante.
� Dal punto di vista genetico nel 70% dei casi è ereditaria e nel 30% rappresenta il frutto di una mutazione de novo.
� Frequenza nella popolazione: 1/5000
� Nelle famiglie in cui siano presenti casi di emofilia è possibile sottoporre le donne all'analisi del DNA (sequenziamento o ricerca RFLP), che si effettua a partire da un normale prelievo di sangue (anche diagnosi prenatale nelle gravidanze a rischio).
� Mutazioni eterogenee (eterogeneiteterogeneitàà allelicaallelica) che si traducono in fenotipi più o meno gravi (eterogeneiteterogeneitàà fenotipicafenotipica). Hot spot nell’introne 22 (nel 50% dei casi).
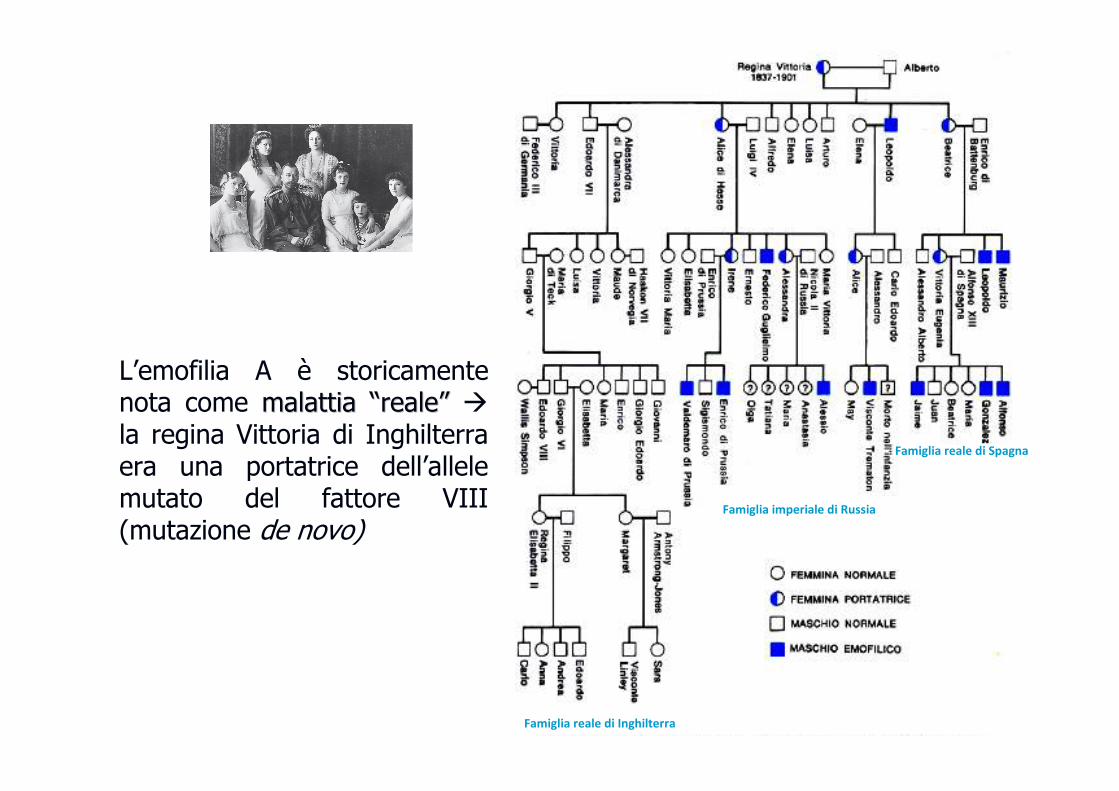
L’emofilia A è storicamente nota come malattia malattia ““realereale”” �
la regina Vittoria di Inghilterra era una portatrice dell’allele mutato del fattore VIII (mutazione de novo)
Famiglia reale di Spagna
Famiglia reale di Inghilterra
Famiglia imperiale di Russia

Emofilia BEmofilia B
� E’ un difetto della coagulazione del sangue trasmesso come carattere legato al cromosoma X e dovuto ad un deficit del fattore IX della coagulazione (gene gene F9F9, Xq27.1)
� Più rara dell’emofilia A: 1/30.000 maschi
�� EterogeneitEterogeneitàà allelicaallelica alta. Anche mutazioni in regioni regolative, assenti nell’emofilia A

Distrofia muscolare di Distrofia muscolare di DuchenneDuchenne
� Frequenza: circa 1/3500 bambini maschi
� Difetto del gene che codifica per la distrofina (Xp21.2 - p21.1), una proteina che si lega alla membrana delle fibre muscolari e stabilisce una connessione tra citoscheletro e matrice extracellulare � preserva l’integrità delle membrane delle cellule muscolari.
� Il gene è molto grande (più di due milioni di basi) con numerose varianti alleliche patologiche (70% delezioni, 30% mutazioni puntiformi) ed espressivitespressivitààvariabilevariabile (fenotipo più grave è la Duchenne, quello meno grave è la Becker)
� Alta percentuale di mutazioni de novo (>30%).
� Caratterizzata da progressiva debolezza muscolare (diagnosi clinica tardiva: 2-6anni) con morte entro i 30 anni
La distrofia muscolare è una malattia degenerativa che colpisce i muscoli. Esistono diversi tipi di distrofia muscolare ma la più frequente in età pediatrica èla forma di Duchenne.
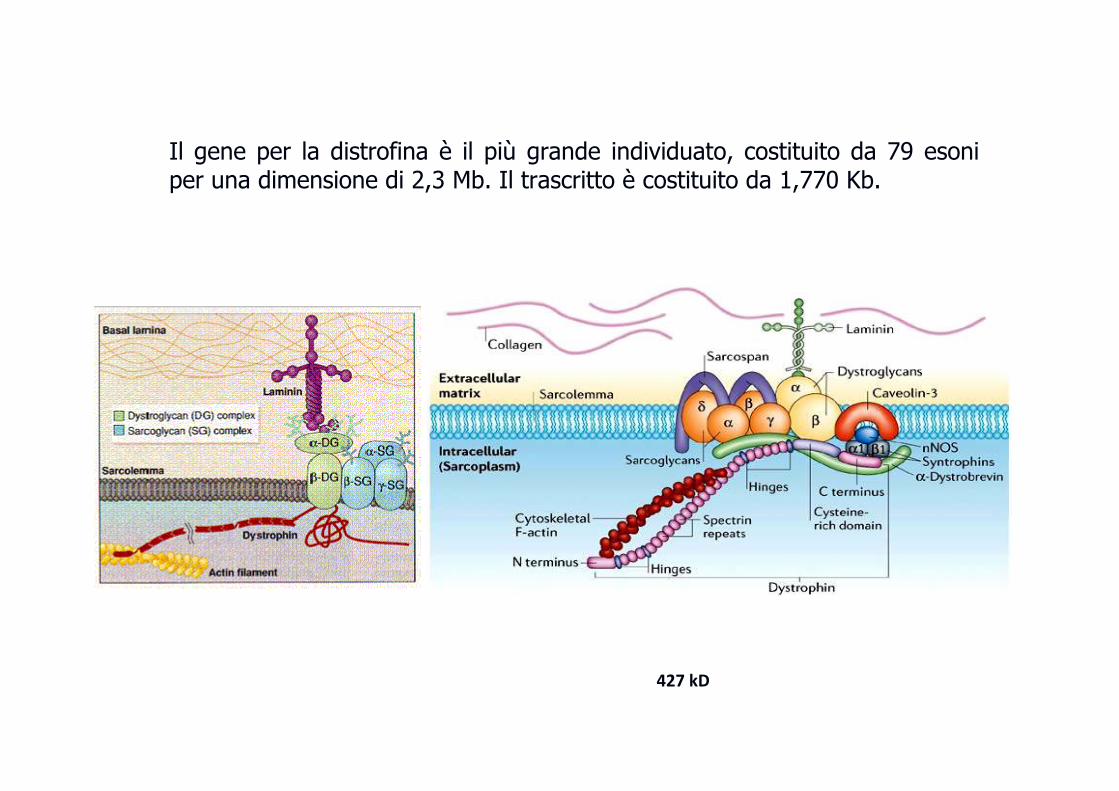
Il gene per la distrofina è il più grande individuato, costituito da 79 esoni per una dimensione di 2,3 Mb. Il trascritto è costituito da 1,770 Kb.
427 kD
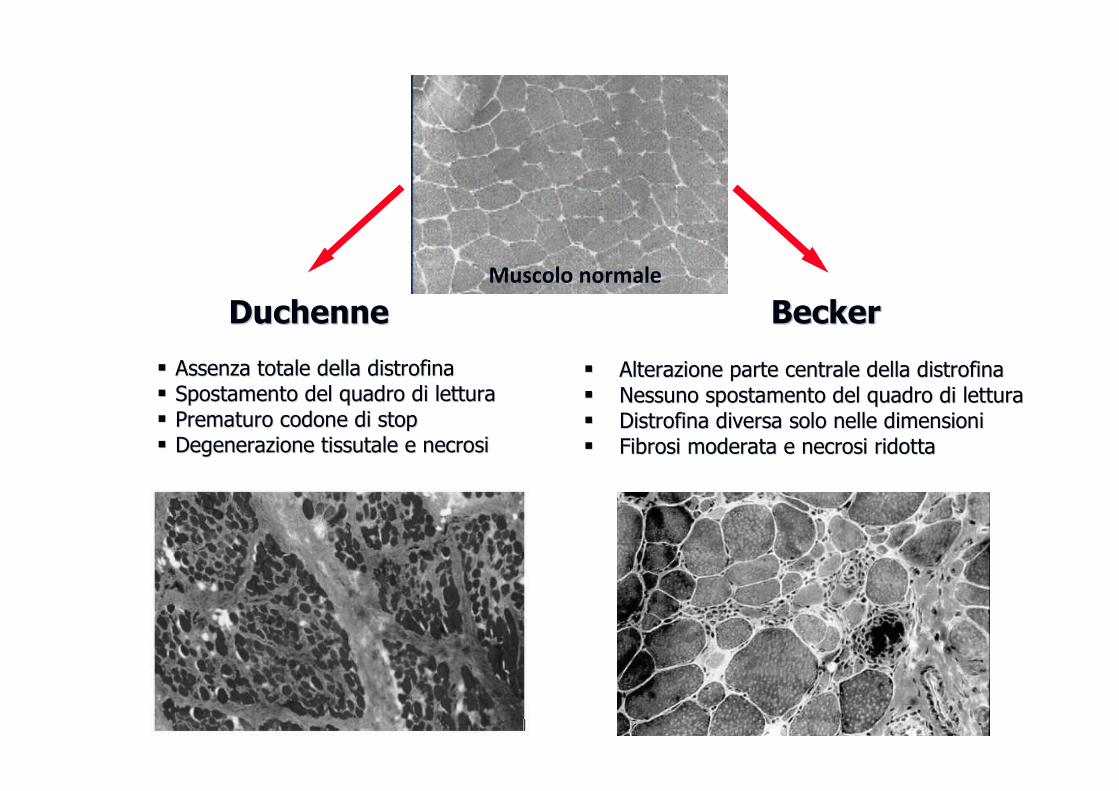
DuchenneDuchenne BeckerBecker
�� Alterazione parte centrale della Alterazione parte centrale della distrofinadistrofina�� Nessuno spostamento del quadro di letturaNessuno spostamento del quadro di lettura�� DistrofinaDistrofina diversa solo nelle dimensionidiversa solo nelle dimensioni�� Fibrosi moderata e necrosi ridottaFibrosi moderata e necrosi ridotta
�� Assenza totale della Assenza totale della distrofinadistrofina�� Spostamento del quadro di letturaSpostamento del quadro di lettura�� Prematuro codone di stopPrematuro codone di stop�� Degenerazione tissutale e necrosiDegenerazione tissutale e necrosi
Muscolo normale
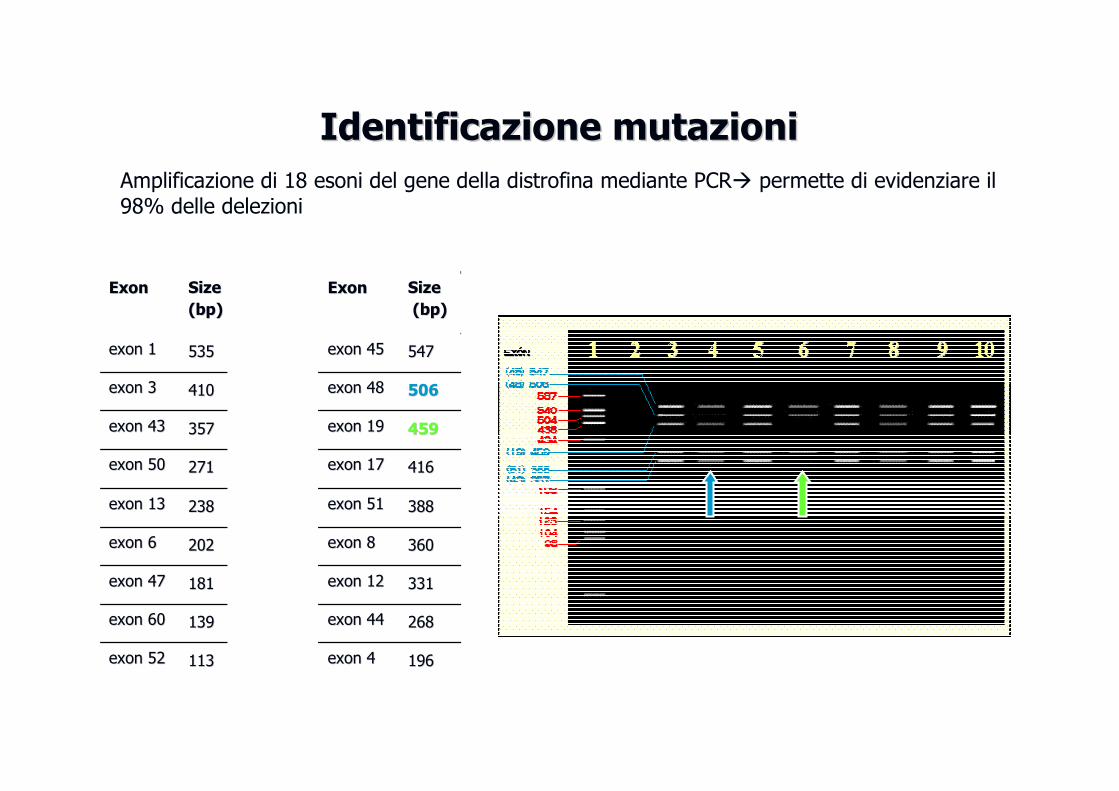
Identificazione mutazioniIdentificazione mutazioni
ExonExon SizeSize
((bpbp))
ExonExon SizeSize
((bpbp))
exon 1exon 1 535535 exonexon 4545 547547
exon 3exon 3 410410 exonexon 4848 506506
exon 43exon 43 357357 exonexon 1919 459459
exon 50exon 50 271271 exonexon 1717 416416
exon 13exon 13 238238 exonexon 5151 388388
exon 6exon 6 202202 exonexon 88 360360
exon 47exon 47 181181 exonexon 1212 331331
exon 60exon 60 139139 exonexon 4444 268268
exonexon 5252 113113 exonexon 44 196196
Amplificazione di 18 esoni del gene della distrofina mediante PCR� permette di evidenziare il 98% delle delezioni

� Malattia dovuta alla mancanza di un'enzima, glucosio-6-fosfato-deidrogenasi (G-6-PDH).
� La forma da carenza di G-6-PDH riguarda circa 400 milioni di persone nel mondo (specialmente in Africa, nell'area tropicale esubtropicale ed in alcune zone del mediterraneo).
� In Italia è molto frequente in Sardegna (mutazioni nel 15% della popolazione) ed in numero più limitato in Sicilia ed in altre regioni meridionali.
�� EterogeneitEterogeneitàà allelicaallelica (circa 400 diverse varianti dell’enzima note) e fenotipicafenotipica
FavismoFavismo

Effetto delle mutazioniEffetto delle mutazioni
� Mutazioni estese (es. frame-shift o delezioni) sono incompatibili con la sopravvivenza nel maschio e molto rare nelle femmine
� La maggior parte delle mutazioni sono missense � proteina instabile o con ridotta attività enzimatica
� La G6PD catalizza la deidrogenazione del G6P e la produzione di NADPH2, usato come cofattore della catalasi per la detossificazione di specie reattive perossidanti come il perossido d’idrogeno (H2O2) e per la riduzione del glutatione. La carenza dell’enzima causa quindi stress ossidativo, particolarmente dannoso per gli eritrociti: emolisi � anemia acuta
� Solitamente si manifesta solo in certe condizioni: esposizione a sostanze particolari, contenute ad es. nelle fave* (da cui il nome) o in alcuni farmaci, che possono scatenare la crisi emolitica
� Diagnosi mediante saggio enzimatico
* Il consumo di fave espone i favici al rischio di crisi emolitiche per via della presenza, all'interno dei semi, di sostanze ossidanti come la vicina e convicina A

Esempi di patologie a trasmissione Esempi di patologie a trasmissione XX--linkedlinked dominantedominante
�� Nefrite ereditariaNefrite ereditaria
�� CondrodisplasiaCondrodisplasia punctatapunctata
Esempi di caratteri/patologie a trasmissione Esempi di caratteri/patologie a trasmissione YY--linkedlinked
�� Retinite pigmentosaRetinite pigmentosa

OMIM, Online Mendelian Inheritance in Man, è una banca dati che contiene
informazioni sui geni umani e sulle malattie genetiche realizzata e mantenuta
dall’NCBI, the National Center for Biotechnology Information. La banca contiene la
descrizione dei geni e delle malattie ad essi associate,i quadri clinici e i riferimenti
bibliografici, oltre a link a sequenze e ad altre risorse web.

Malattie monogeniche- Tipo di trasmissione -
Localizzazione cromosomica- Ref. OMIM
Corea di Huntington -1 / 20000- Autosomica dominante- Cromosoma 4-
143100
Fibrosi cistica - 1 / 2000-4000 - Autosomica recessiva -Cromosoma 7 -
219700
Anemia falciforme (HbS)- Autosomica recessiva- Cromosoma 11-
141900.0243
Talassemia -Autosomica recessiva- Cromosoma 11 catene beta;
cromosoma 16 catene alfa -Alfa: 141800 Beta: 141900
Fenilchetonuria- 1 / 10000 nati vivi- Autosomica recessiva- Cromosoma
12 -261600
Emofilia A- 1 / 5000-10000 nei maschi - X-linked recessiva- Cromosoma
X -306700
Miopatia di Duchenne- 1 / 6000 nei maschi -X-linked recessiva-
Cromosoma X- 310200

MIM ID #310200
MUSCULAR DYSTROPHY, DUCHENNE TYPE; DMD
Alternative titles; symbols DUCHENNE MUSCULAR DYSTROPHY , MUSCULAR
DYSTROPHY, PSEUDOHYPERTROPHIC PROGRESSIVE, DUCHENNE TYPE
Gene map locus: Xp21.2Table of Contents
MIM #310200
Text
Description
Clinical Features
Other Features
Inheritance
Cytogenetics
Mapping
Molecular Genetics
Diagnosis
Clinical Management
Population Genetics
Animal Model
Clinical Synopsis







