

BASI GENETICHE DELLA MALATTIA DI ALZHEIMER
Valentina Gatta
Professore Associato
Genetica Medica Università “G. D’Annunzio” Chieti
Centro Studi dell’Invecchiamento (Ce.SI) Fondazione “G. D’Annunzio” Chieti
0871-541456 [email protected]

Alois Alzheimer
• La malattia di Alzheimer è stata descritta per la prima volta nel 1906 dal neuropatologo Alois Alzheimer

Malattia di
Alzheimer (AD)
DR. ALOIS
ALZHEIMER
La malattia di Alzheimer è la forma piùfrequente di demenza,
colpisce circa il 5% delle personeultrasessantacinquenni circa il 10% di
quelle oltre gli 85 anni
La malattia ha un inizio subdolo: lepersone cominciano a dimenticare alcunecose per arrivare al punto in cui nonriconoscono nemmeno i familiari ed hannobisogno di aiuto anche per le attivitàquotidiane più semplici.

Oltre 100 cause di Demenza
A . Degenerative
• Malattia di Alzheimer
• Malattia di Pick
• Malattia di Huntington
• Atrofia Frontotemporale senza corpi di Pick
• Malattia a corpi di Lewy
• Malattia di Parkinson
• Malattia di Wilson
• Paralisi Sopranucleare Progressiva
• Degenerazione Spinocerebellare
• Degenerazione Corticobasale
• Afasia Progressivea
• Demenza Semantica
• Atrofia corticale posteriore
B. Vascolari
• Multi-infartuale
• Malattia di Binswanger
• Vasculiti
• Ematoma Subdurale
• Infarto Strategico
• Ipoperfusione
• Postemorragica
C. Miste vascolari e degenerative
D. Miscellanea (malattie
respiratorie ostruttive; sleep
apnea; radiazioni; dialisi;
privazione di sonno; ipossia)

•36 milioni i malati nel mondo, di cui 6 milioni in Europa e 1 milione in Italia•incremento in Italia di 150.000 nuovi casi all'anno•aumento progressivo delle demenze, circa 66 milioni nel 2030 e quasi 116 milioni nel 2050•costi sociali e sanitari pari all’uno per cento del PIL Mondiale, più di 600 miliardi di dollari.
*stime dell’Alzheimer's Disease International (Adi)
Impatto sociale ed economico

Una delle patologie a più grave impatto sociale del mondo-ampia e crescente diffusione nella popolazione-limitata e comunque non risolutiva efficacia delle terapie disponibili-enormi risorse necessarie per la suagestione (sociali, emotive, organizzative ed economiche), che ricadono in gran parte sui familiari dei malati
Impatto sociale

Il 90% delle persone con problemi di demenza sono assistite dalle famiglie-a domicilio
Impatto sociale

La famiglia rappresenta il principale supporto assistenziale per i malati affetti dalla demenza di tipo Alzheimer e da altra patologia cronica.-ricerca di un equilibrio all’interno della famiglia, sin dalla diagnosi, si stabilisce un equilibrio che si evolve e si aggiusta con la progressione e della malattia-malattia del caregiver-aggravamento dei disturbi comportamentali.
Aiuti possibili:-informazione sulla natura della demenza-rete dei servizi-supporto delle associazioni
Impatto socialeLa famiglia del malato di Alzheimer
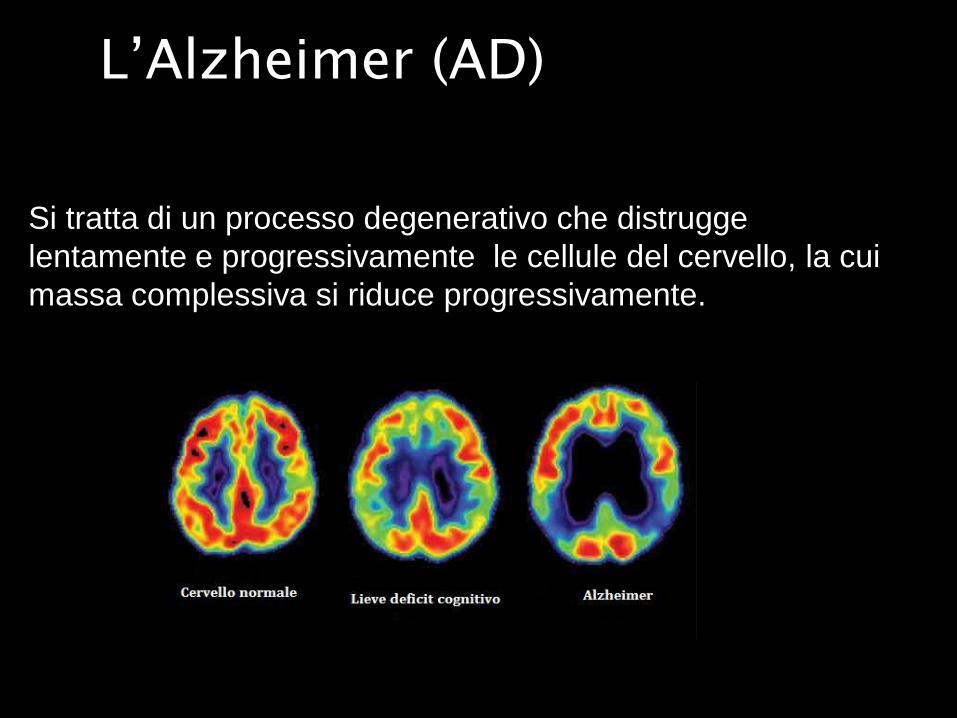
L’Alzheimer (AD)
Si tratta di un processo degenerativo che distrugge
lentamente e progressivamente le cellule del cervello, la cui
massa complessiva si riduce progressivamente.

AREE ENCEFALICHECOINVOLTE
I. Gangli della base contenenti acetilcolina, neurotrasmettitore importante per memoria e apprendimento.
II. Ippocampo, essenziale per la conservazione della memoria.
III.Corteccia cerebrale, dove vengono elaborati pensiero e linguaggio.

Si tratta di un disturbo acquisito, e con base organica, delle funzioni intellettive che
sono state in precedenza acquisite: memoria (a breve e lungo termine) e almeno
una tra pensiero astratto, capacità critica, linguaggio, orientamento spazio
temporale, con conservazione dello stato di coscienza vigile
Essa inizia con l’indebolimento della memoria, in particolare per i fatti recenti; le sensazioni si affievoliscono, l’attenzione è labile e alla lunga
impossibile; la volontà è incerta e senza vigore; i movimenti sono lenti e imprecisi

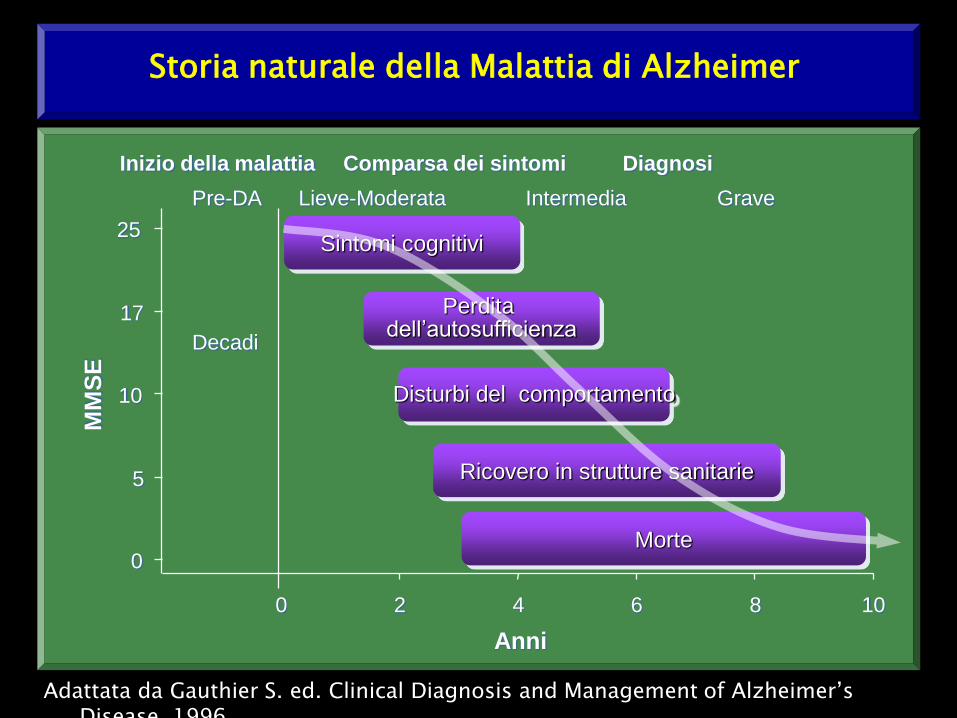
Storia naturale della Malattia di Alzheimer
Adattata da Gauthier S. ed. Clinical Diagnosis and Management of Alzheimer’s Disease. 1996.
0
5
10
17
25
0 2 4 6 8 10
Anni
Sintomi cognitivi
Perdita dell’autosufficienza
Disturbi del comportamento
Ricovero in strutture sanitarie
Morte
Decadi
Pre-DA Lieve-Moderata Intermedia Grave
Inizio della malattia Comparsa dei sintomi Diagnosi
MM
SE

PLACCHE DI AMILOIDE:
depositi amorfi extracellulari di proteina -amiloide (A)
sotto forma di placche
GROVIGLI NEUROFIBRILLARI:
intreccio intracellulare
di neurofibrille formate da proteina TAU anomala
Patologia della malattia di Alzheimer

La Proteina Precursore dell’Amiloide (APP) è una proteina posta in parte
all’interno della membrana cellulare e in parte all’esterno. Essa è molto
importante per la crescita, la sopravvivenza e la riparazione della cellula.

APP
• APP può essere tagliata in frammenti funzionali da tre enzimi che operano tagli proteolitici: la α-secretasi e la β-secretasi in un primo momento e successivamente la γ-secretasi.
• Attraverso due tagli successivi operati prima dall'α-secretasi e poi dall'γ-secretasi, viene prodotto un peptide innocuo chiamato p3.
• La β-secretasi opera un taglio differente che, in seguito al successivo taglio da parte della γ-secretasi, porta alla produzione (pathway amiloidogenico) di due peptidi di 40 e 42 aminoacidi, chiamati beta-amiloide (Aβ 1-40 e Aβ 1-42): il secondo (Aβ 1-42) è considerato il più tossico a livello neuronale.
• Nei soggetti sani il processo di degradazione della APP sembra essere operato principalmente dalla α-secretasi.
• Per motivi non totalmente chiariti, nei soggetti malati l'enzima che interviene sull'APP non è l'α-secretasi ma la β-secretasi, con una larga produzione di proteina beta-amiloide.
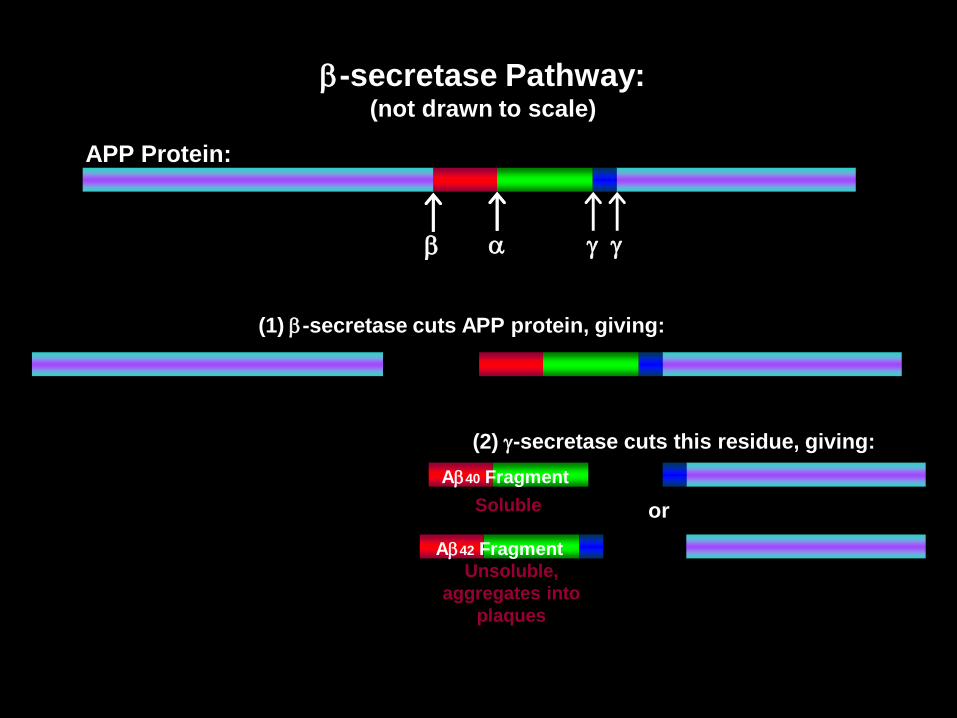
a g g
APP Protein:
(1) -secretase cuts APP protein, giving:
(2) g-secretase cuts this residue, giving:
or
A40 Fragment
Soluble
A42 Fragment
Unsoluble,
aggregates into
plaques
-secretase Pathway:(not drawn to scale)

Il precursore dell’amiloide
Queste proteasi sono sotto il controllo di due altre proteasi: le preseniline 1 e 2
Mutazioni nelle preseniline, a-, -, e g-secretasi o nella APP stessa determinano
l’accumulo extracellulare di A che sono causa di malattia.
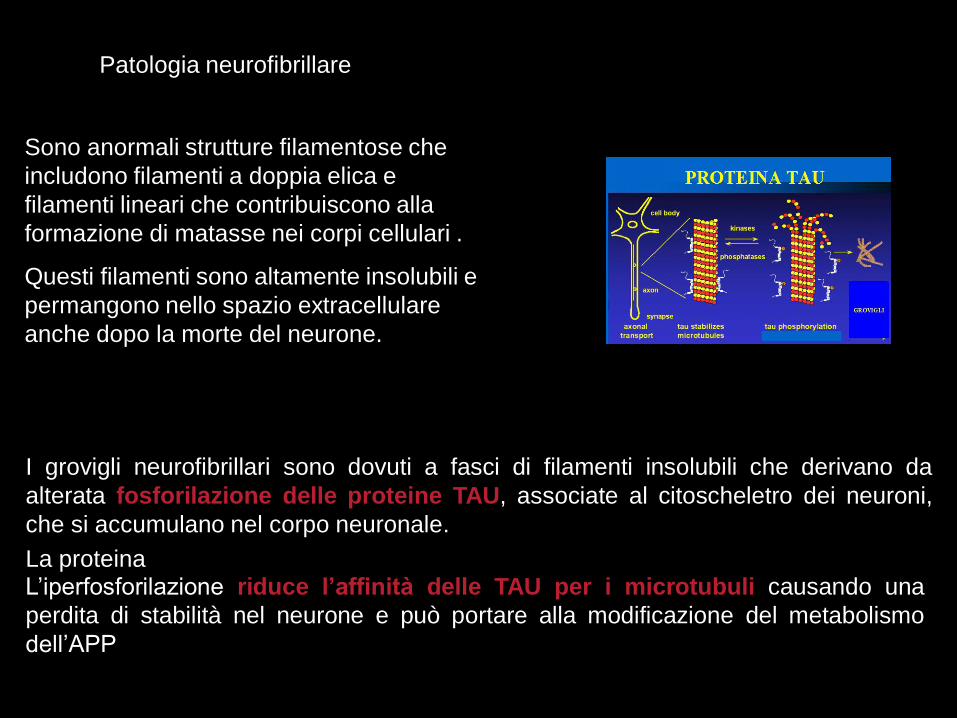
I grovigli neurofibrillari sono dovuti a fasci di filamenti insolubili che derivano da
alterata fosforilazione delle proteine TAU, associate al citoscheletro dei neuroni,
che si accumulano nel corpo neuronale.
La proteinaL’iperfosforilazione riduce l’affinità delle TAU per i microtubuli causando una
perdita di stabilità nel neurone e può portare alla modificazione del metabolismo
dell’APP
Sono anormali strutture filamentose che
includono filamenti a doppia elica e
filamenti lineari che contribuiscono alla
formazione di matasse nei corpi cellulari .
Questi filamenti sono altamente insolubili e
permangono nello spazio extracellulare
anche dopo la morte del neurone.
Patologia neurofibrillare

The amyloid cascade hypothesis:
…DEMENZA…

Eziopatogenesi
•Fattori di rischio:
– Genetici
– Età
– Differenze di genere
–Storia di traumi– Fattori modificabili


•Forma sporadica (75%)
•Forma associata a S. di Down (1%)
•Forma familiare a insorgenza tardiva (20%)
•Forma familiare a insorgenza precoce (5%)

GENETICA
•La Genetica si occupa:
• della struttura
•della trasmissione
•dell’espressione dell’informazione ereditaria.
DNA

Struttura1953


Elmenti base del DNAA=TC=G

GENETICA
•La Genetica si occupa:
• della struttura
•della trasmissione
•dell’espressione dell’informazione ereditaria.
DNA

Trasmissione dei caratteri• Mendel (1856-1863)
Leggi della ereditabilità
dei caratteri
Processo mediante il quale caratteri
specifici vengono trasmessi dai
genitori ai figli.
l'ereditarietà di questi caratteri è
controllata da determinati fattori,
chiamati geni.

Trasmissione• L’insieme delle
caratteristiche genetiche rappresenta il GENOTIPO
• Unione di due patrimoni, uno di origine paterna ed uno di origine materna

Cariotipo

Cromosomi e geni
• Sui cromosomi di ogni individuo sono presenti i geni, ognuno in una specifica posizione (locus)
• Ogni gene determina direttamente o indirettamente uno o più fenotipi


Lo stratagemma del codice genetico
AGTGCTGATCGTAGCTAGCTAGCTAGCTAG
AGT-GCT-GAT-CGT-AGC-TAG-CTA-GCT-AGC-TAG
aa1 aa2 aa3 aa4 aa5 aa6 aa7 aa8 aa9 aa10
Proteina X
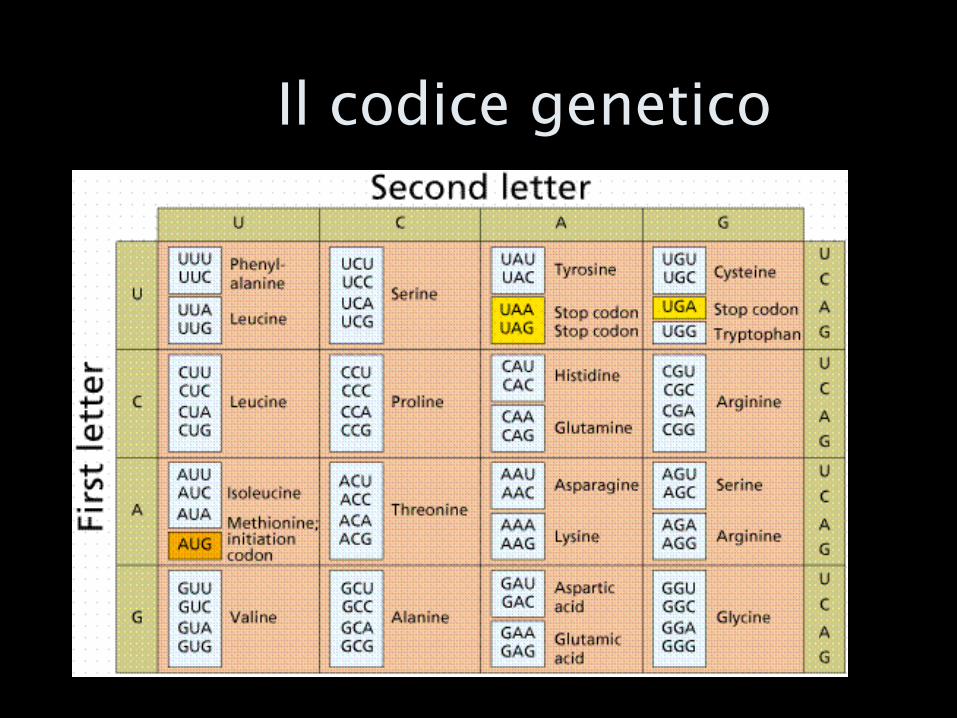
Il codice genetico

Siamo fatti di pochi ingredienti:
acqua, sali, zuccheri, grassi, proteine:
LA NOSTRA INDIVIDUALITA' DIPENDE SOLO DALLE
PROTEINE

Il ruolo delle proteine
• Le proteine prodotte dai geni agiscono in un individuo determinando, insieme all’ambiente:– L’aspetto fisico
– Il metabolismo
– Le capacità cognitive
– La capacità di reagire a stimoli esterni
• Tale ruolo è esercitato attraverso 3 funzioni:– Strutturale
– Ormonale
– Enzimatica


Come possono i geni fare tutto?
• Una catena di processi molecolari che conducono da un singolo gene ad un particolare carattere ereditario
• I geni devono governare la sintesi delle proteine mediante un processo detto

Cosa succede se altero la sequenza di un gene??
LE MUTAZIONI
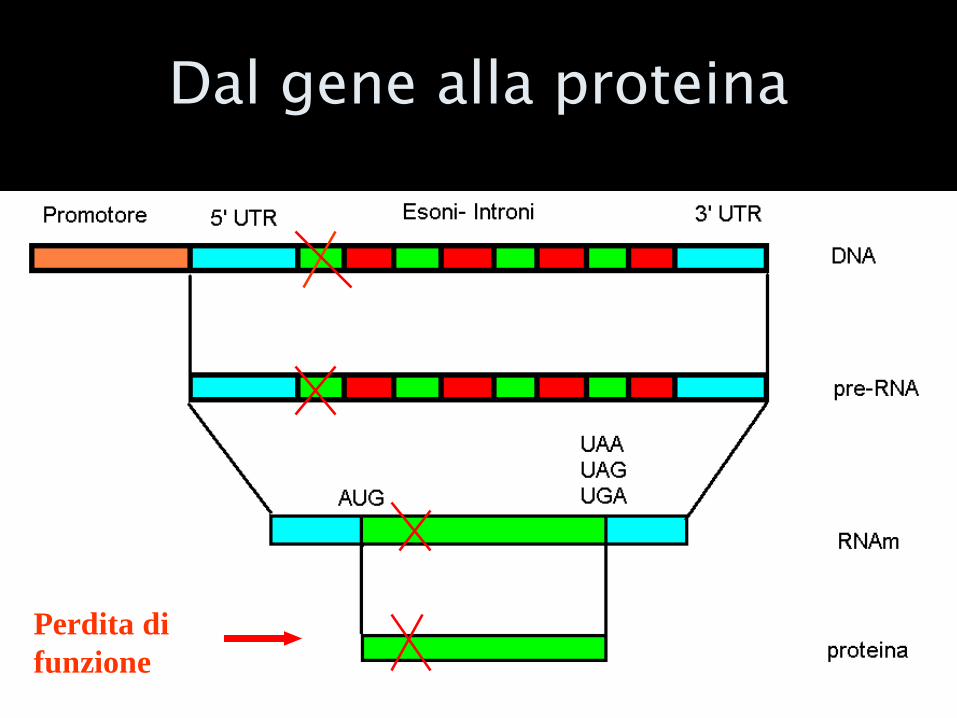
Dal gene alla proteina
Perdita di
funzione

Cos’è una mutazione?



Mutazioni
• Cambi nella normale sequenza del DNA
• Sede:
– Germinali (ereditabili)
– Somatiche (non ereditabili)
• Origine:
– Spontanea (errori di replicazione o riparazione del DNA)
– Indotta (da agenti fisici o chimici)

Agenti mutageni




Effetti delle mutazioni
• Mutazioni letali– Provocano la morte del 100% dei portatori prima della
pubertà
• Mutazioni subletali– Portano a morte più del 50% (ma non il 100%) dei portatori
prima della pubertà
• Mutazioni neutrali– Non modificano la salute dell’individuo in senso negativo o
positivo
• Mutazioni vantaggiose– Sono favorevoli per la specie, aumentando la fitness
riproduttiva dei portatori

VARIAZIONE DEI CARATTERI
•
…nel bene…nel male

Darwin (1809-1882)
• Esistenza di una ampiavariabilità
• Ereditarietà dei caratteri tra genitori e figli
• Selezione: in un determinato ambiente alcune forme hanno più successo di altre

Dal genotipo al fenotipo

Patologia Genetica
• Patologia Monogenica:– malattie dovute a mutazioni di un singolo
gene
• Patologia Poligenica:– malattie dovute a mutazioni o polimorfismi di
più geni, generalmente con presenza di una componente ambientale
• Patologia Cromosomica:– malattie dovute ad alterazioni numeriche o
strutturali del cariotipo

Le malattie a trasmissione mendeliana: Autosomiche Dominanti,
Recessive e X-linked

Malattie a trasmissione autosomica dominante
• Sono quelle in cui l’allele mutato prevale su quello sano, ed è pertanto dominante
– Omozigote dominante AA = malato
– Omozigote recessivo aa = sano
– Eterozigote Aa = malato

Alberi genealogici con trasmissione
di malattie autosomiche dominanti
• Gli affetti hanno di norma un genitore affetto
• In media il 50% dei figli di un affetto sono affetti
• Non vi è salto di generazione

Nanismo acondroplasico
• Trasmissione autosomica dominante a penetranza completa
• Incidenza: 1/25.000• 7/8 dei casi risultati di
nuove mutazioni• Locus: 4p16• Gene: FGFR3• Mutazione: 90% dei
casi transizione G-A con cambio di una glicina in arginina (mutazione da acquisto di funzione)

Malattie a trasmissione autosomica recessiva
• Sono quelle in cui l’allele sano prevale su quello malato, che è quindi recessivo
– Omozigoti dominanti AA= sani
– Omozigoti recessivi aa = malati
– Eterozigoti Aa = portatori sani
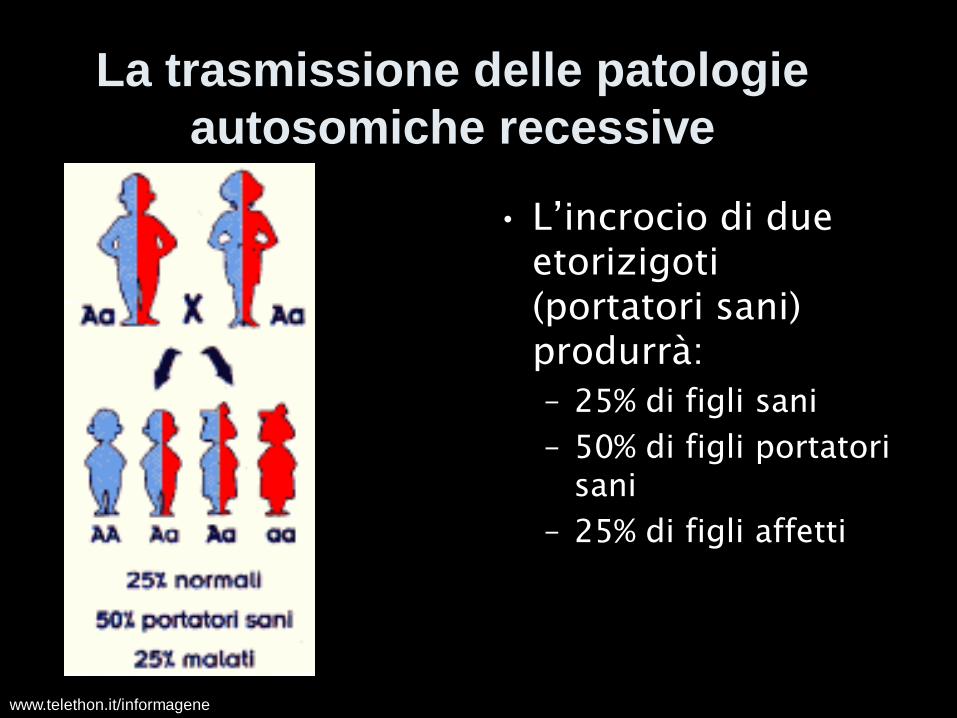
• L’incrocio di due etorizigoti (portatori sani) produrrà:
– 25% di figli sani
– 50% di figli portatori sani
– 25% di figli affetti
www.telethon.it/informagene
La trasmissione delle patologie
autosomiche recessive

Alberi genealogici con trasmissione di malattie autosomiche recessive
• Una coppia di portatori sani avrà un rischio del 25% di avere figli malati
• In genere presente il salto di generazione
• Frequente riscontro di consanguineità dei genitori

Altre malattie autosomiche recessive
• Emocromatosi ereditaria (1/350-1/2000)– Accumulo di ferro nell’organismo– Danni a carico di fegato, pancreas e cuore
• Anemia Falciforme (1/400-1/600 afroamericani)– Anemia cronica con tipiche deformazioni degli eritrociti
• Sordità bilaterale (1/1.1700)
• Atrofia Muscolare Spinale (1/10.000)– Morte dei motoneuroni del midollo spinale– Paralisi progressive con età di esordio e gravità variabili
• Albinismo (1/30.000)

Patologia Genetica
• Patologia Monogenica:– malattie dovute a mutazioni di un singolo
gene
• Patologia Complessa:– malattie dovute ad alterazioni di più geni, con
presenza di una componente ambientale
• Patologia Cromosomica:– malattie dovute ad alterazioni numeriche o
strutturali del cariotipo

• Il fenotipo è un carattere quantitativo, derivante dalla interazione tra fattori genetici e ambientali

Caratteri complessi
• Schizofrenia
• Anoressia/Bulimia
• Autismo
• Infarto
• Tumori
• …..

Patologia Genetica
• Patologia Monogenica:– malattie dovute a mutazioni di un singolo
gene
• Patologia Poligenica:– malattie dovute a mutazioni o polimorfismi di
più geni, generalmente con presenza di una componente ambientale
• Patologia Cromosomica:– malattie dovute ad alterazioni numeriche o
strutturali del cariotipo

Patologia cromosomica


Tipi diversi di morbo di Alzheimer
•Forma familiare a insorgenza tardiva (20%)
•Forma familiare a insorgenza precoce (5%)
•Forma sporadica (75%)
•Forma associata a S. di Down (1%)

Forme familiari
• esordio precoce
• esordio tardivo

Forma esordio precoce familiare (5%)(AD1, AD3, AD4)
• Forma mendeliana eredità autosomica dominante
• Diagnosi in famiglie con più casi della malattia che si manifestano prima dei 65 anni. I sottotipi (AD1, AD4, AD3) si possono distinguere solo con un test genetico.

Cause genetiche

AD Familiare ad esordio tardivo dopo i 65 anni (20%)
– Patologia multigenica con diversi loci di suscettibilità
– Rischio di ricorrenza familiare: 20%
– Clinicamente indistinguibili da quella sporadica

•Presente in tutti i Down dopo i 40 anni
•Aumentata espressione del gene APP (precursore della amiloide), situato sul cromosoma 21

Forma Sporadica
• Forma sporadica (75%): assenza di storia familiare
• Ipotesi patogenetica multifattoriale:
– Età
– Predisposizione genetica (ApoE)
– Agenti ambientali
• Sostanze tossiche
• Traumi

AD esordio tardivo
• ApoE principale gene candidato nell’AD

Apo E
• ApoE (Apolipoproteina E) : lipoproteina coinvolta
nel metabolismo del colesterolo, trasporto e rilascio di lipidi tra differenti cellule o tessuti
• Implicata nella riparazione neuronale
• Presente in tre isoforme ε3, ε2, ε4

Rimozione dell’amiloide
Ruolo dell’ApolipoproteinaE (ApoE)
Proteina plasmatica implicata nel trasporto del colesterolo
Nel SNC interviene nei meccanismi di crescita e riparazione dopo un danno
Nell’AD la ApoE è legata alle placche di A e ai grovigli neurofibrillari
ApoE inibisce la formazione di fibrille di A, ma perde efficacia se associata ai lipidi
I peptidi amiloidi interagiscono direttamente con ApoE legandovisi e potrebberoessere poi trasportati all’interno delle cellule neuronali via recettori per ApoE
Nei cervelli di soggetti sani ApoE si lega e sequestra A in modo più efficiente chein quelli dei malati di AD
La deplezione di colesterolo comporta una riduzione nella produzione e nel rilasciodi A
Aggregazione promossa per aggiunta di ioni metallici, ApoE facilita o attenua ilprocesso a seconda del metallo

Correlazione ApoE ed AD

AD esordio tardivo :

Nuove tecnologie


Sequenziamento del DNA
• Tecnica che ci permette di determinare il preciso susseguirsi di basi del DNA.
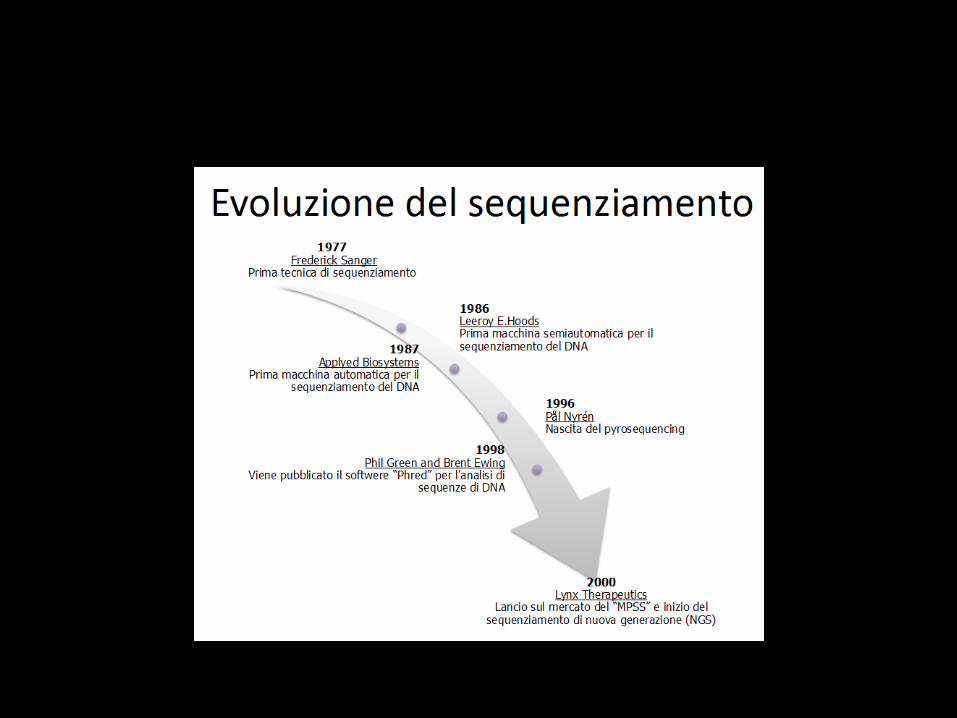

Next gen sequencing
• Tecnologia che è in grado di sequenziare a bassi costi ed alta processività .
• Genoma umano in 10 giorni.
• Progetto Genoma è durato 10 anni


Il sequenziamento di interi genomi: progetto genoma
• Il sequenziamento di interi genomi si è basato sulla costruzione di genoteche che rappresentavano la collezione completa di cloni di DNA :clonaggio
• Progetto Genoma è durato 10 anni
• Essendo il genoma umano di 3 Χ 109 basi, sarebbero necessarie 107 sequenze di 300 basi

• 2005: prima piattaforma di NGS commercializzata è stata l’FLX Genome Sequencer della 454 Life Sciences (Branfrd, Connecticut, USA) ora proprietà del gruppo Roche
• 2007 dal Genome Analyzer I della Illumina (San Diego, California, USA)
• SOLiD della Applied Biosystems (Foster City, California, USA).



PGM
• La nuova tecnologia del PGM si differenzia in modo drastico dai sequenziatori di seconda generazione:
– materiale semiconduttore
– leggere le sequenze di DNA misurando le variazioni di pH che si generano quando un nuovo nucleotide viene aggiunto al filamento
sintetizzato.

Elimina fonti di errore
• Basi modificate
• Basi fluorescenti
• Laser detection
• Cascata di amplificazione
enzimatica
• Basi non naturali
• Sintesi difettosa
• Tempi lunghi

Chip 314 Chip 316 Chip 318
10 Mb 100 Mb 1 Gb
1.3 milioni 6.2 milioni 11.1 milioni
Chip 314 Chip 316 Chip 318
10 Mb 100 Mb 1 Gb
1.3 milioni 6.2 milioni 11.1 milioni
MicroArray
• High Density MouseOneArray Whole GenomeDNA microarray (PhalanxBiotech)
• 31,802 trascritti

Early and Sustained Altered Expression of
Aging-Related Genes in Young 3xtg-AD Mice
• Valentina Gatta1,2, Alberto Granzotto3, Marco D’Aurora2,4, • Liborio Stuppia1,2, and Stefano L. Sensi3,4,5,6*
• 2 Functional Genetics Unit, Center of Excellence on Aging (Ce.S.I.), Chieti, Italy
• 3 Molecular Neurology Unit, Center of Excellence on Aging (Ce.S.I.), Chieti, Italy
• 4 Department of Neuroscience and Imaging, “G. D’Annunzio” University
» +

SCOPO
Evidenziare, mediante MicroArray, i
profili di espressione genica “age-
dependent”, di ippocampi di modelli
murini AD-like a 3 e 12 mesi di età
rispetto a topi WT della stessa età
Valutare i network genici e i pathway che
sono coinvolti sia nell’invecchiamento che
nella patologia neurodegenerativa di
Alzheimer

Modello animale (3xTG)
Human mutant APP (swedish mutation)
Human Mutant PS1
Human mutant tau (P301L)
Overespressione dei transgeni ristretta al SNC
Deposizione di peptidi di A a 4-6 mesi di età
Danneggiati la trasmissione sinaptica e la long-termpotentiation a 6-9 mesi di età
Formazione di aggregati di forma alterata e detection ditau fosforilata nell’ippocampo a 12-15 mesi di età
U.O. Neurologia Molecolare - Ce.S.I.
Prof. Sensi S.L.

Confronto tra ippocampi: 3xTG a 3 mesi di età vs WT
a 3 mesi di età
3xTG a 12 mesi di età vsWT a 12 mesi di età
Aging fisiologico WT a 12 mesi di età vs WT
a 3 mesi di età
Femmine 3xTG-AD (n=4)
Femmine WT (n=4)
Disegno sperimentale
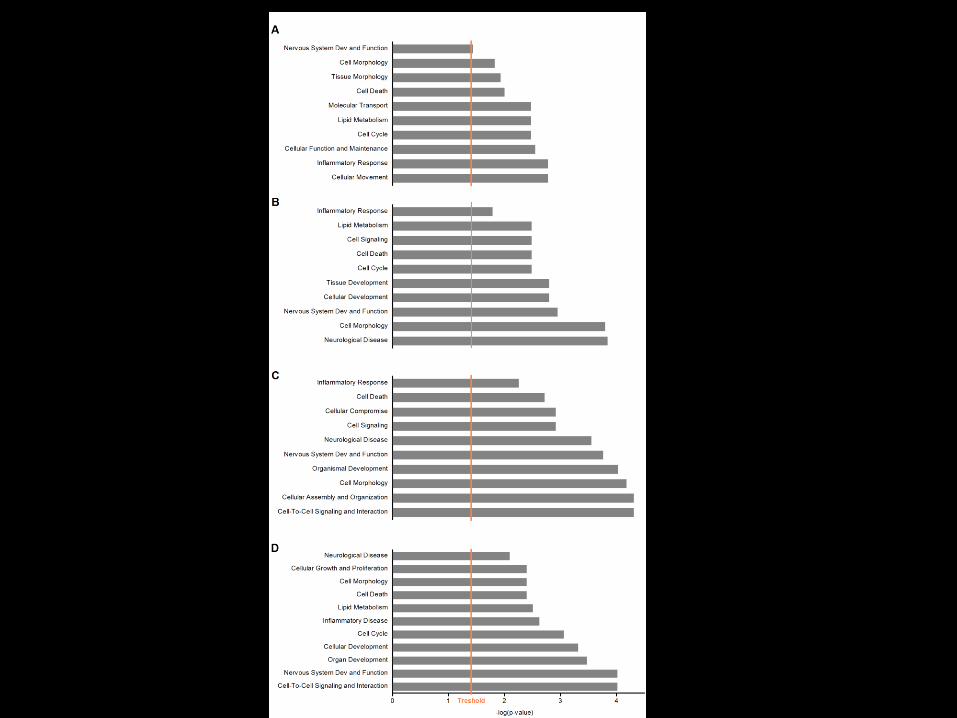
L’analisi dei dati ottenuti dai è stata eseguita mediante clustering gerarchico(Cluster 3.0) e visualizzata con il software TreeView (Stanford University Labs)

A
B
C
D
L’analisi Cluster ha evidenziato 4gruppi genici differenzialmenteespressi• A 37 geni sovra-espressi con
l’invecchiamento e sotto-espressi nei 3xTG a 3 e 12 mesirispetto ai WT
• B 62 geni sovra-espressi sianell’invecchiamento che nei3xTG a 3 mesi e sotto-espressinei 3xTG a 12 mesi
• C 193 geni sovra-espressi intutte le condizioni
• D 77 geni sotto-espressi sianell’invecchiamento che nei3xTG a 3 mesi e sovra-espressinei 3xTG a 12 mesi
WT
12vs
WT
3
3xT
G3vs
WT
3
3xT
G12vs
WT
12

Conclusione
• Alterazioni a livello della espressione genica sono evidenziabili in età precoce.
• Il profilo di espressione in un modello animale giovane è simile a quello di età avanzata
• Biomarker predittivi

La genetica dell’AD
Tanzi et al. 2012 Oct

Eziopatogenesi
•Fattori di rischio:
– Genetici
– Età
– Differenze di genere
–Storia di traumi

Guardando il futuro
Nei prossimi 40 anni il numero di persone affette di AD si triplica.


Leggere nel futuro







