
1
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 1
The use of slides from Prof. Piero Ugliengo’s lectures is acknowledged
Slides adapted from the lecture given at MSSC2007 by Cesare Pisani
Chimica Fisica dei Materiali e laboratorio
Modellizzazione di difetti locali in
materiali cristallini
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 2
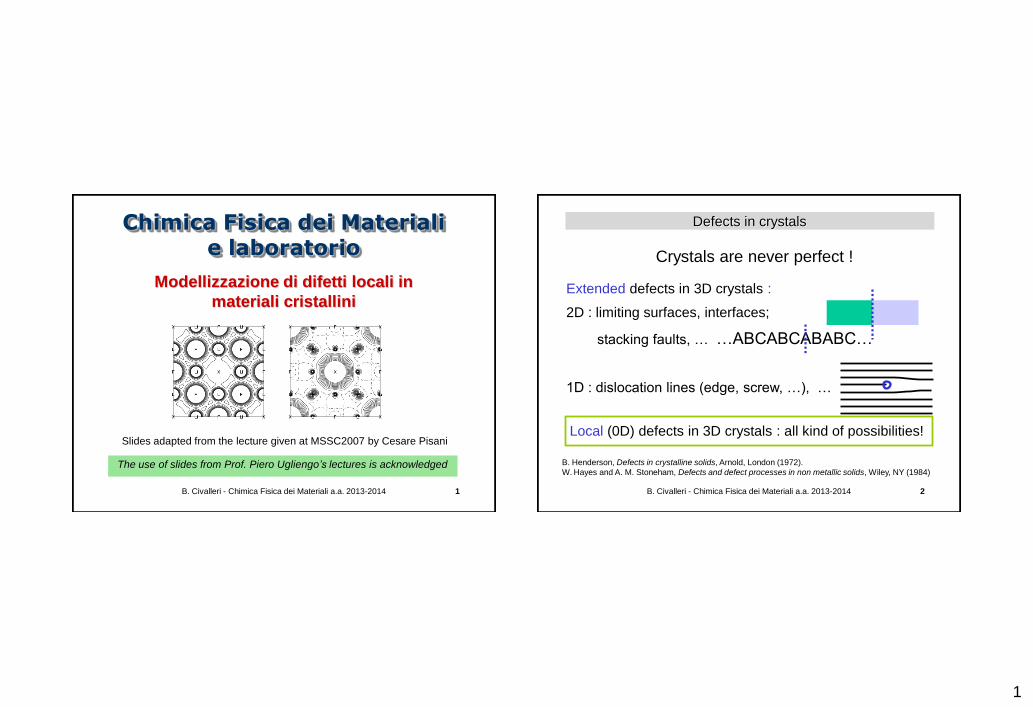
Defects in crystals
Crystals are never perfect !
Extended defects in 3D crystals :
2D : limiting surfaces, interfaces;
stacking faults, … …ABCABCABABC…
1D : dislocation lines (edge, screw, …), …
Local (0D) defects in 3D crystals : all kind of possibilities!
B. Henderson, Defects in crystalline solids, Arnold, London (1972).
W. Hayes and A. M. Stoneham, Defects and defect processes in non metallic solids, Wiley, NY (1984)
2
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 3
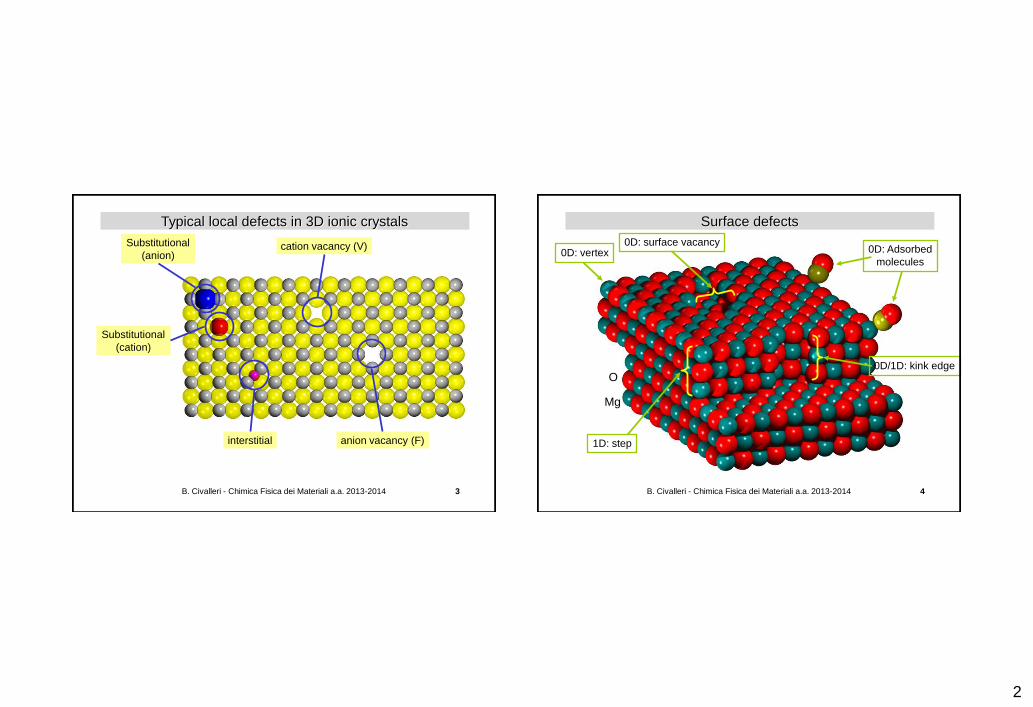
Typical local defects in 3D ionic crystals
cation vacancy (V)
anion vacancy (F) interstitial
Substitutional
(anion)
Substitutional
(cation)
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 4
Surface defects
0D: vertex
1D: step
0D/1D: kink edge
0D: Adsorbed
molecules
0D: surface vacancy
Mg
O

3
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 5
QM simulation of local defects: fundamentals
Sf
+ +
+ R S'(q) + P {E(q)}
defect zone
• The perfect host crystal assumption:
Sf (the host system) is a perfect (and known ) crystalline structure
• The locality assumption:
Sf and S' structures coincide outside the defect zone (finite and small)
• The QM problem:
(i) determine the equilibrium geometry of S'(q) (ii) describe the change of the electronic structure in the defect zone (iii) evaluate E(q) (a finite difference between infinite energies!!)
• The self-embedding condition:
without defects, all features of S'(q) should coincide with those of Sf
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 6
Local defects in crystals: a classification of QM strategies
Modelling of the defect and solution technique are inter-related
aspects which characterize the approach to the QM problem.
A possible classification of strategies is as follows:
B. “Periodic strategies”:
Sf (and possibly S') is modelized as a perfect crystal
b1) Supercell approach
b2) Multi-level (P-ONIOM) technique
b3) Embedding techniques (perturbative, Green-functions)
b4) -Partition (group-function) techniques
A. “Non-Periodic (cluster) strategies”:
Both Sf and S' are simulated as a finite cluster
a1) Bare (bond-saturated) clusters
a2) Dressed clusters (extra terms to simulate environmental effects)
a3) Multi-level (ONIOM) technique
4
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 7
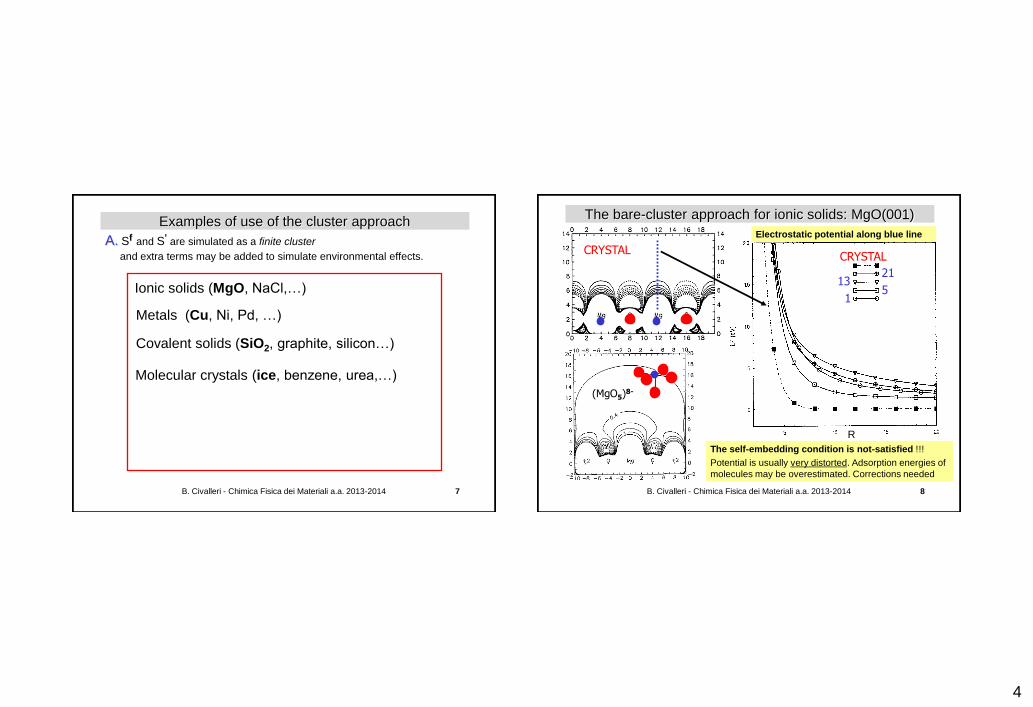
Examples of use of the cluster approach
A. Sf and S' are simulated as a finite cluster
and extra terms may be added to simulate environmental effects.
Ionic solids (MgO, NaCl,…)
Covalent solids (SiO2, graphite, silicon…)
Molecular crystals (ice, benzene, urea,…)
Metals (Cu, Ni, Pd, …)
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 8
The bare-cluster approach for ionic solids: MgO(001)
CRYSTAL
1
CRYSTAL
5
21 13
Electrostatic potential along blue line
(MgO5)8-
The self-embedding condition is not-satisfied !!!
Potential is usually very distorted. Adsorption energies of
molecules may be overestimated. Corrections needed
R
5
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 9
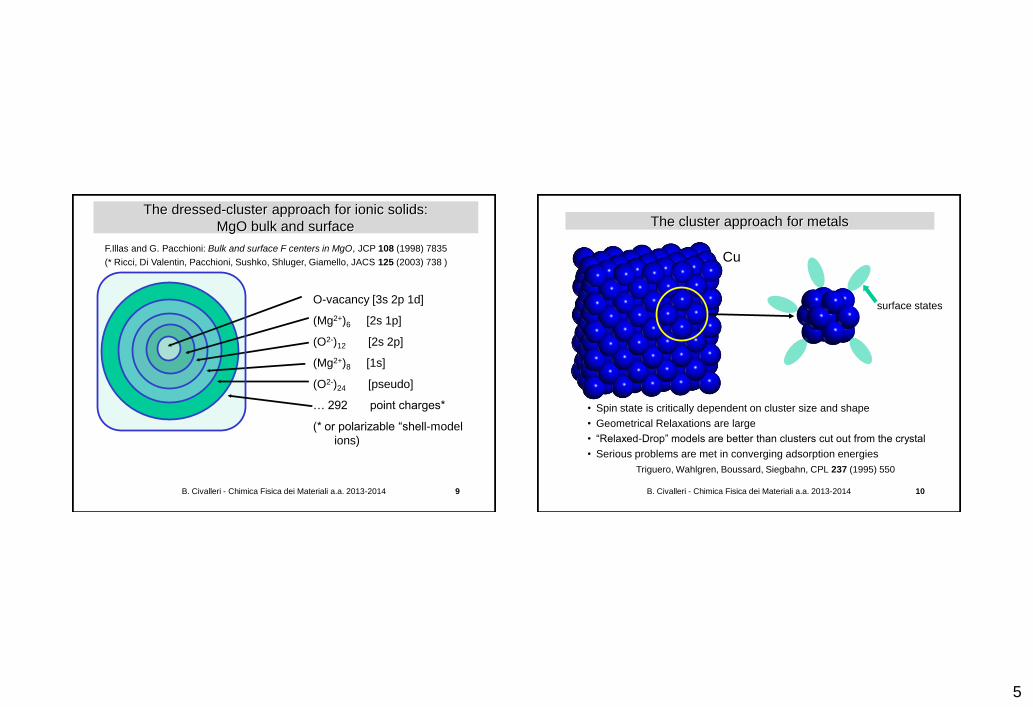
The dressed-cluster approach for ionic solids:
MgO bulk and surface
F.Illas and G. Pacchioni: Bulk and surface F centers in MgO, JCP 108 (1998) 7835
(* Ricci, Di Valentin, Pacchioni, Sushko, Shluger, Giamello, JACS 125 (2003) 738 )
O-vacancy [3s 2p 1d]
(Mg2+)6 [2s 1p]
(O2-)12 [2s 2p]
(Mg2+)8 [1s]
(O2-)24 [pseudo]
… 292 point charges*
(* or polarizable “shell-model
ions)
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 10
The cluster approach for metals
Cu
• Spin state is critically dependent on cluster size and shape
• Geometrical Relaxations are large
• “Relaxed-Drop” models are better than clusters cut out from the crystal
• Serious problems are met in converging adsorption energies
Triguero, Wahlgren, Boussard, Siegbahn, CPL 237 (1995) 550
surface states

6
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 11
The cluster approach for molecular crystals
• Geometry resulting from the cut
will change dramatically upon relaxation
• Molecules at the border will move to maximize the
intermolecular interactions (H-bond)
• Optimized structures are no longer representative of the
crystal to which they initially belong
• Arbitrarly shaped clusters may develop huge dipole
moment absent in the periodic system
Bussolin, Casassa, Pisani, Ugliengo, J. Chem. Phys., 108, 9516 (1998).
unsatisfied
H-bond
unsatisfied
H-bond B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 12
The saturated-cluster approach for covalent solids
• N° atoms grows fast with cluster size • Large number of terminal H atoms
• Hard to enforce crystal memory into the cluster structure
Consider quartz as a prototype of covalent material. Silicon and other
semiconductors all belongs to this category
7
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 13
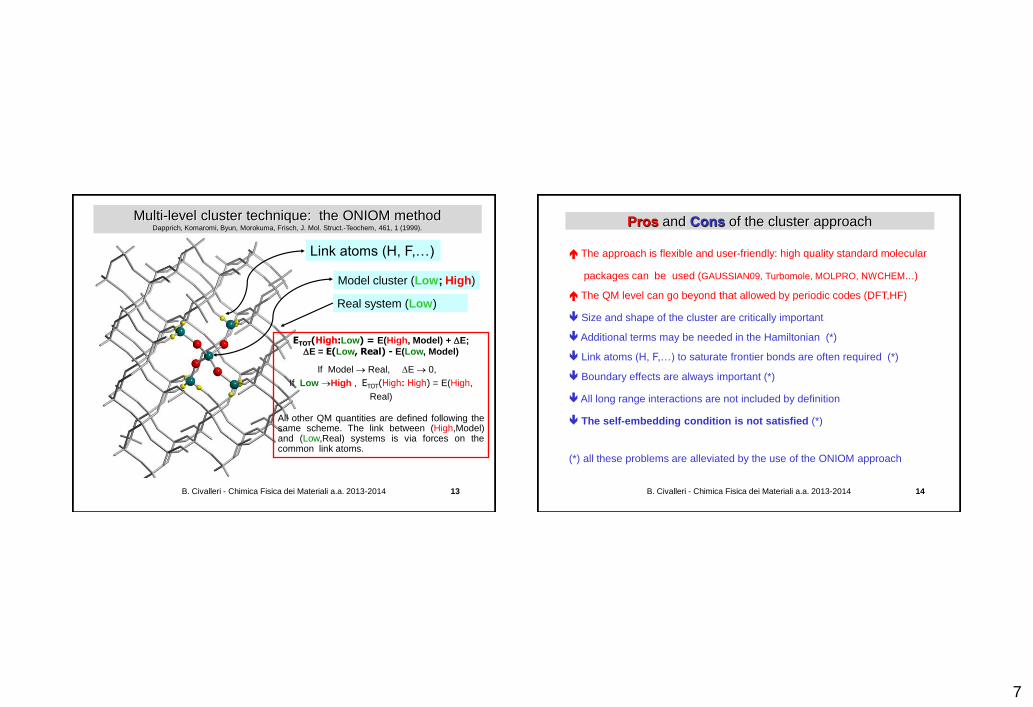
Multi-level cluster technique: the ONIOM method Dapprich, Komaromi, Byun, Morokuma, Frisch, J. Mol. Struct.-Teochem, 461, 1 (1999).
Link atoms (H, F,…)
Model cluster (Low; High)
Real system (Low)
ETOT(High:Low) = E(High, Model) + E;
E = E(Low, Real) - E(Low, Model)
If Model Real, E 0,
If Low High , ETOT(High: High) = E(High,
Real)
All other QM quantities are defined following the same scheme. The link between (High,Model) and (Low,Real) systems is via forces on the common link atoms.
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 14
Pros and Cons of the cluster approach
The approach is flexible and user-friendly: high quality standard molecular
packages can be used (GAUSSIAN09, Turbomole, MOLPRO, NWCHEM…)
The QM level can go beyond that allowed by periodic codes (DFT,HF)
Size and shape of the cluster are critically important
Additional terms may be needed in the Hamiltonian (*)
Link atoms (H, F,…) to saturate frontier bonds are often required (*)
Boundary effects are always important (*)
All long range interactions are not included by definition
The self-embedding condition is not satisfied (*)
(*) all these problems are alleviated by the use of the ONIOM approach
8
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 15
Local defects in crystals: “Periodic” approaches
B. The defect as an impurity of the host perfect crystal:
Sf is treated as a perfect crystalline structure
and S' is treated (in principle) at the same level of accuracy.
B1. The supercell (SC) approach
B2. The Periodic Multilevel (P-ONIOM) approach
B3. The embedded cluster Green-function approach
B4. The -partition approach
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 16
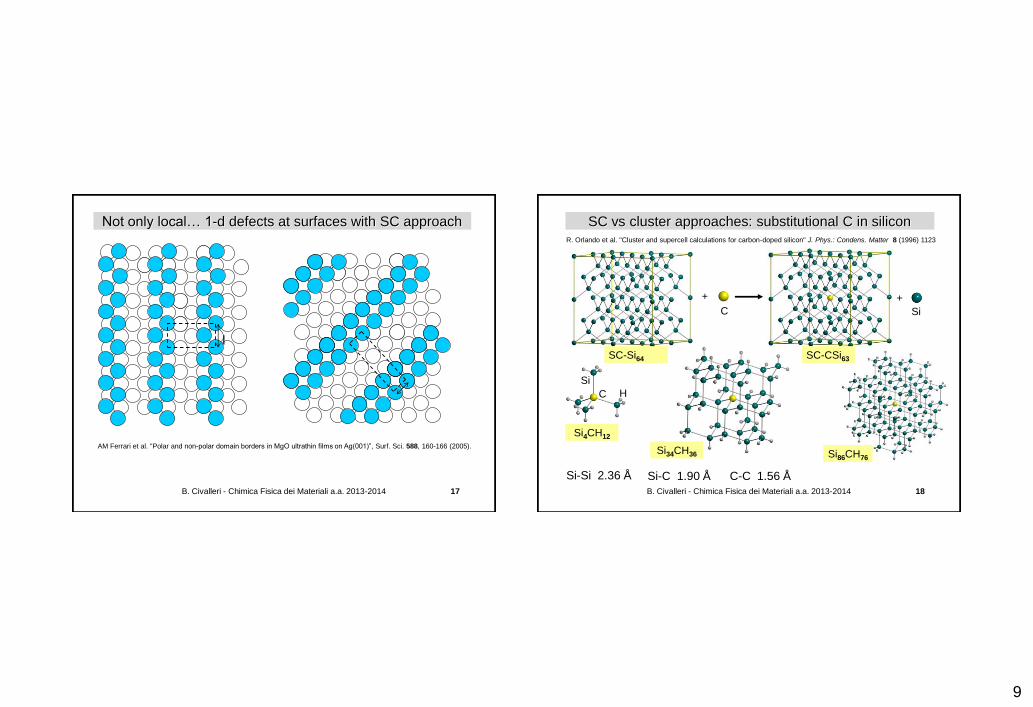
Local defects in crystals: the super-cell (SC) approach
Sf perfect host
n1=2, n2=2 n1=1, n2=1
S'SC
defective host
E = lim(n1, n2, n3) [ E(S'SC) – n1 n2 n3 E(Sf) ] + E(P) – E(R)
9
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 17
Not only local… 1-d defects at surfaces with SC approach
l
AM Ferrari et al. "Polar and non-polar domain borders in MgO ultrathin films on Ag(001)”, Surf. Sci. 588, 160-166 (2005).
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 18
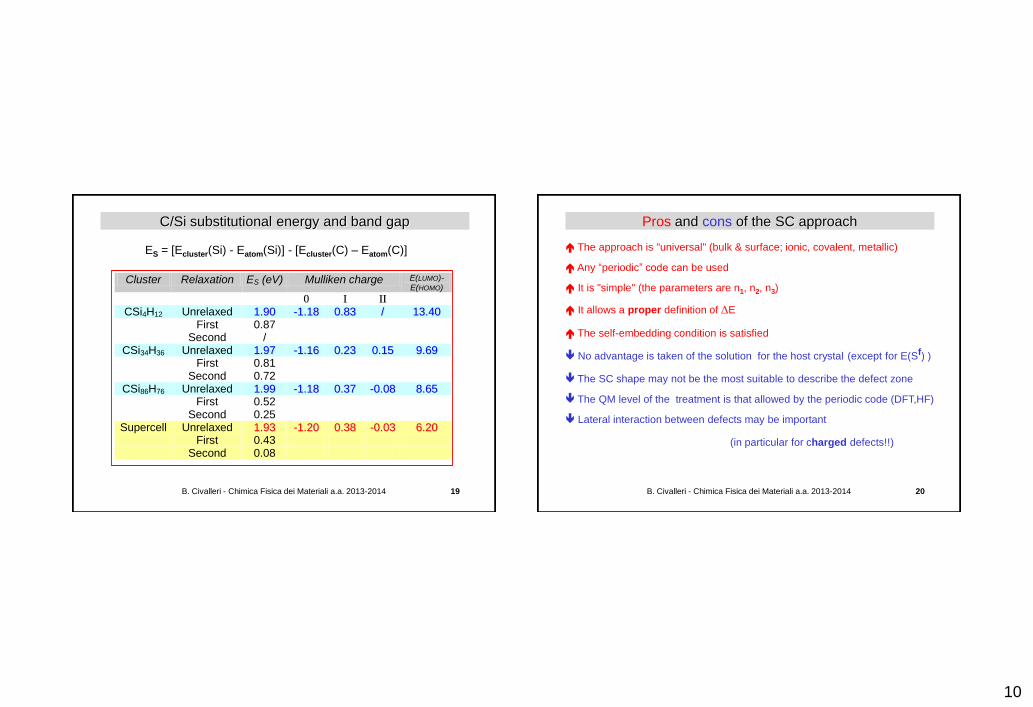
SC vs cluster approaches: substitutional C in silicon R. Orlando et al. "Cluster and supercell calculations for carbon-doped silicon" J. Phys.: Condens. Matter 8 (1996) 1123
SC-Si64 SC-CSi63
C Si
+ +
Si86CH76 Si34CH36
Si4CH12
Si-Si 2.36 Å Si-C 1.90 Å C-C 1.56 Å
C
Si H
10
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 19
C/Si substitutional energy and band gap
ES = [Ecluster(Si) - Eatom(Si)] - [Ecluster(C) – Eatom(C)]
Cluster Relaxation ES (eV) Mulliken charge E(LUMO)-E(HOMO)
00 II IIII
CSi4H12 Unrelaxed 11..9900 --11..1188 00..8833 // 1133..4400 First 0.87 Second /
CSi34H36 Unrelaxed 11..9977 --11..1166 00..2233 00..1155 99..6699 First 0.81 Second 0.72
CSi86H76 Unrelaxed 11..9999 --11..1188 00..3377 --00..0088 88..6655 First 0.52 Second 0.25
Supercell Unrelaxed 11..9933 --11..2200 00..3388 --00..0033 66..2200 First 0.43 Second 0.08
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 20
The approach is "universal" (bulk & surface; ionic, covalent, metallic)
Any “periodic” code can be used
It is "simple" (the parameters are n1, n2, n3)
It allows a proper definition of E
The self-embedding condition is satisfied
No advantage is taken of the solution for the host crystal (except for E(Sf) )
The SC shape may not be the most suitable to describe the defect zone
The QM level of the treatment is that allowed by the periodic code (DFT,HF)
Lateral interaction between defects may be important
(in particular for charged defects!!)
Pros and cons of the SC approach

11
O Be Li O
Axial defect
Defect-defect interactions?
Low density defective system High density defective system
[Li]0 center in beryllium oxide
Supercell Etot(BeO) EBeO
S32 (2 2 2) -1435.23633 -89.70227
S48 (2 2 3) -2152.85450 -89.70227
S64 (2 2 4) -2870.47266 -89.70227
S72 (3 3 2) -3229.28175 -89.70227
S108 (3 3 3) -4843.92262 -89.70227
S144 (3 3 4) -6458.56349 -89.70227
S180 (3 3 5) -8073.20437 -89.70227
S216 (3 3 6) -9687.84524 -89.70227
S252 (3 3 7) -11302.48612 -89.70227
S128 (4 4 2) -5740.94533 -89.70227
S192 (4 4 3) -8611.41799 -89.70227
S256 (4 4 4) -11481.89066 -89.70227
S300 (5 5 3) -13455.34061 -89.70227
Etot(BeOLi) E
-1427.83826 0.25818
-2145.45400 0.26061
-2863.07115 0.26162
-3221.89975 0.24211
-4836.53959 0.24314
-6451.18019 0.24341
-8065.82080 0.24368
-9680.46139 0.24396
-11295.10206 0.24417
-5733.56656 0.23888
-8604.03731 0.24079
-11474.50982 0.24095
-13447.96104 0.23968
Axial defect E = (Edef + EBe) – (Eper + ELi) UHF
EBeo= -89.70227 ; EBe= -14.56948 ; ELi= -7.42959 Energies in Hartree
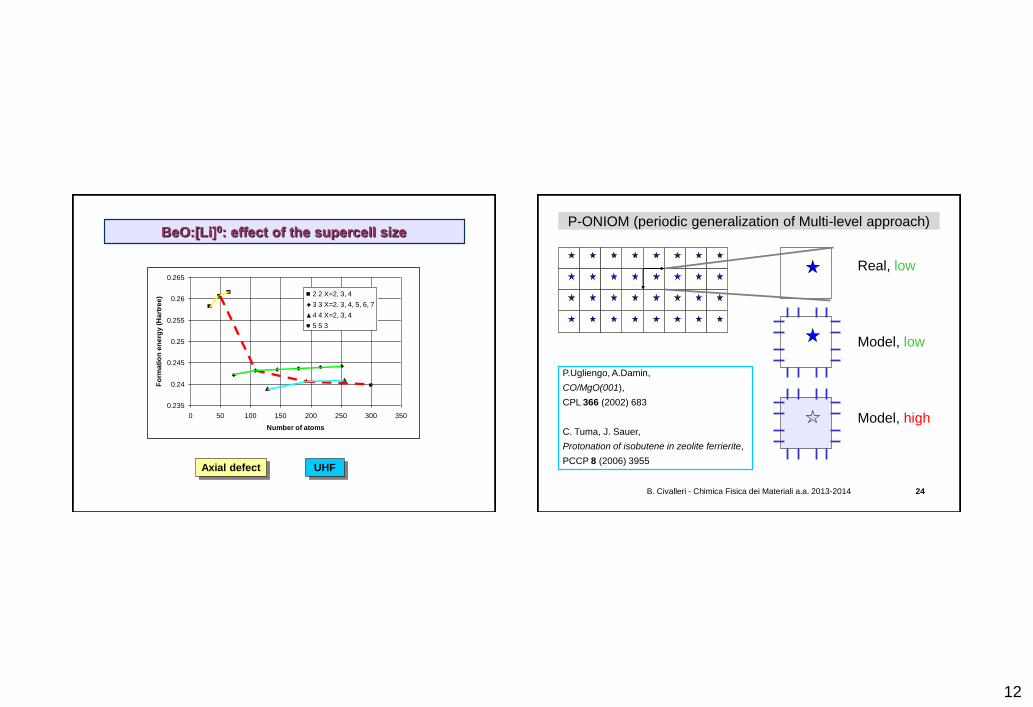
BeO:[Li]0: effect of the supercell size
12
0.235
0.24
0.245
0.25
0.255
0.26
0.265
0 50 100 150 200 250 300 350
Number of atoms
Fo
rma
tio
n e
ne
rgy
(H
art
ree
) 2 2 X=2, 3, 4
3 3 X=2, 3, 4, 5, 6, 7
4 4 X=2, 3, 4
5 5 3
BeO:[Li]0: effect of the supercell size
Axial defect UHF
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 24
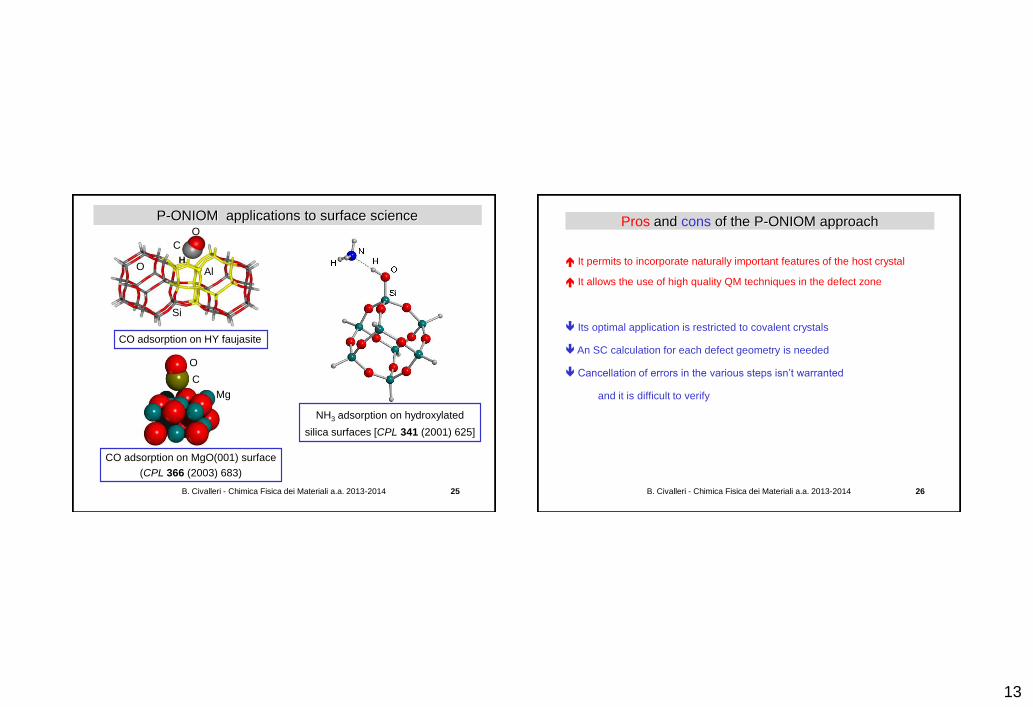
P-ONIOM (periodic generalization of Multi-level approach)
P.Ugliengo, A.Damin,
CO/MgO(001),
CPL 366 (2002) 683
C. Tuma, J. Sauer,
Protonation of isobutene in zeolite ferrierite,
PCCP 8 (2006) 3955
Real, low
Model, low
Model, high
13
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 25
P-ONIOM applications to surface science
NH3 adsorption on hydroxylated
silica surfaces [CPL 341 (2001) 625]
C
O
Mg
CO adsorption on MgO(001) surface
(CPL 366 (2003) 683)
C
O
Al H
O
Si
CO adsorption on HY faujasite
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 26
It permits to incorporate naturally important features of the host crystal
It allows the use of high quality QM techniques in the defect zone
Its optimal application is restricted to covalent crystals
An SC calculation for each defect geometry is needed
Cancellation of errors in the various steps isn’t warranted
and it is difficult to verify
Pros and cons of the P-ONIOM approach

14
B. Civalleri - Chimica Fisica dei Materiali a.a. 2013-2014 27
CONCLUSIONS
The study of defects in crystals (especially surface defects)
is an important field of application of periodic codes
The super-cell approach is currently the preferred choice
The use of “hybrid” (Multi-Level) models is often recommended,
to allow a high-quality treatment of the defect zone
Prospective developments include the use of -Partition techniques,
based on a Wannier-function description of the ground-state
wavefunction of the host crystal







