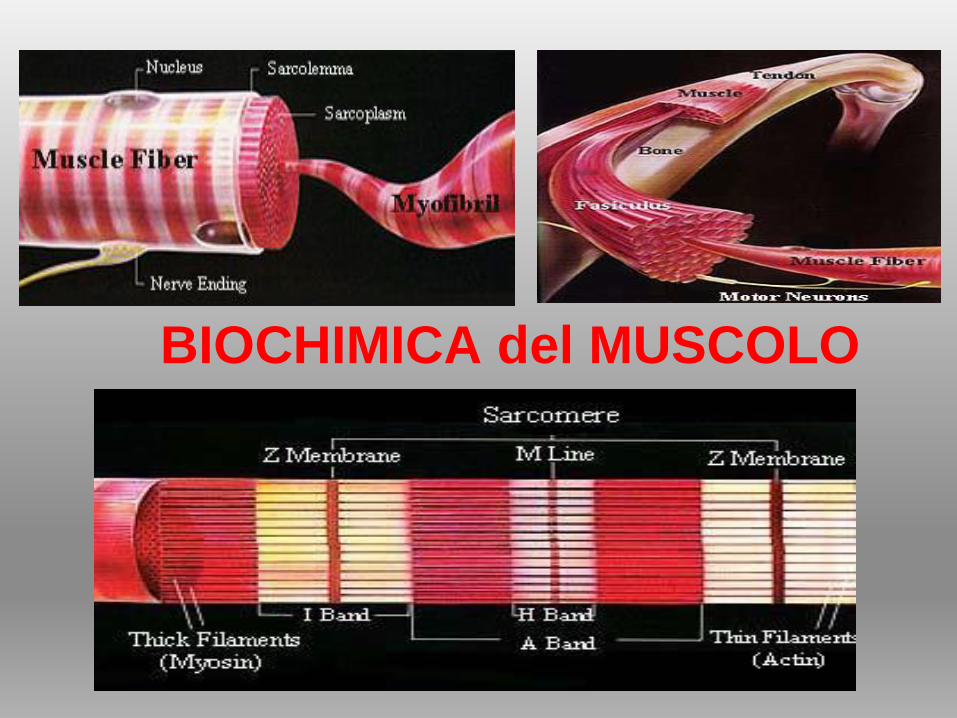
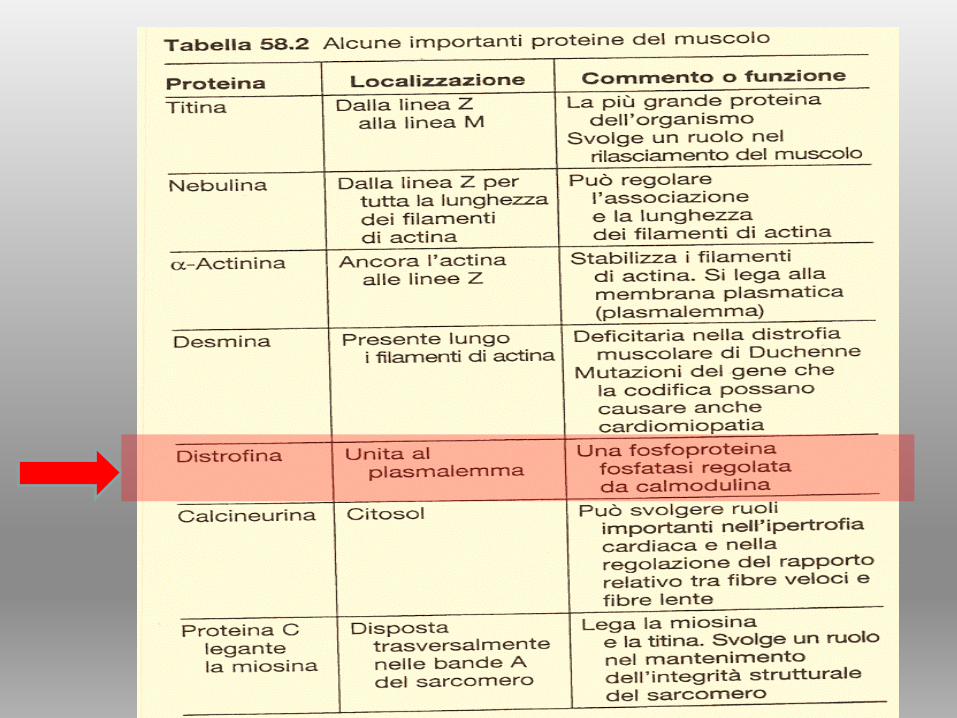
BIOCHIMICA del MUSCOLO

TESSUTO MUSCOLARE
• 40% massa corporea permette sia i movimenti del corpo nell’insieme che quelli di singole parti.
• Principale tessuto utilizzatore di ATP
• Fonte energetica prevalente: acidi grassi liberi (tx adiposo), utilizza anche scorte di glicogeno e corpi chetonici.
• Contrazione muscolare dipendente dalla presenza di proteine contrattili tra le quali ACTINA e MIOSINA e di proteine strutturali, ATP e Ca2+.
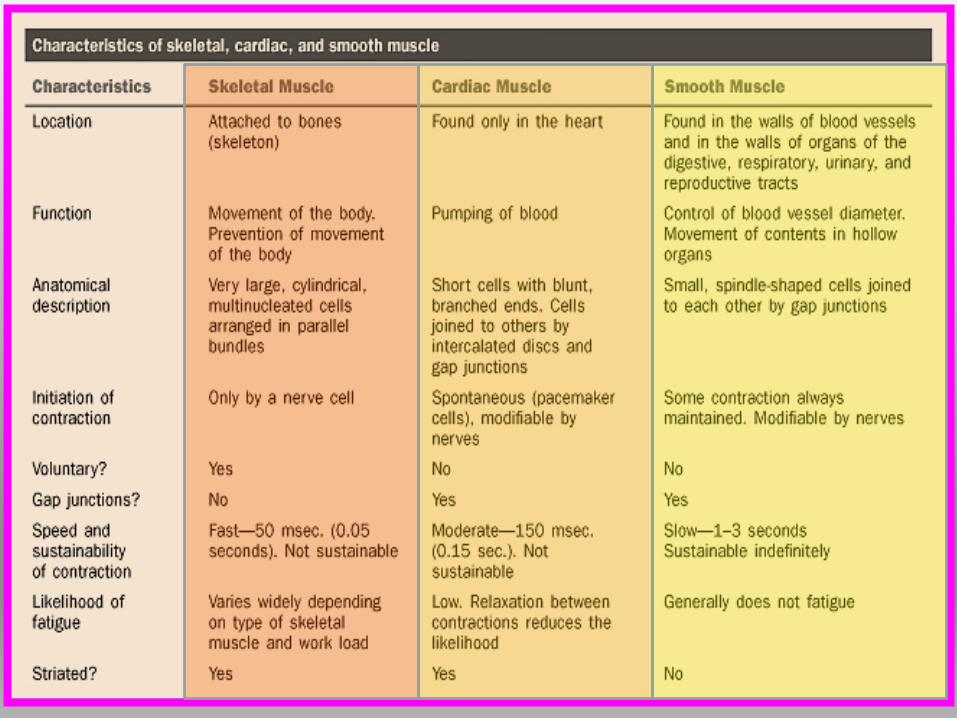
MUSCOLO STRIATO
SCHELETRICO: muscolo
volontario dipende dal
sistema nervoso. Presenta
striature trasversali chiare
e scure: BANDE.
MUSCOLO STRIATO
CARDIACO: simile
scheletrico presenza di
bande, contrattilità ritmica
ed automatica, indipendente
da volontà.
MUSCOLO LISCIO:
muscolo involontario.
Non mostra striature
trasversali.
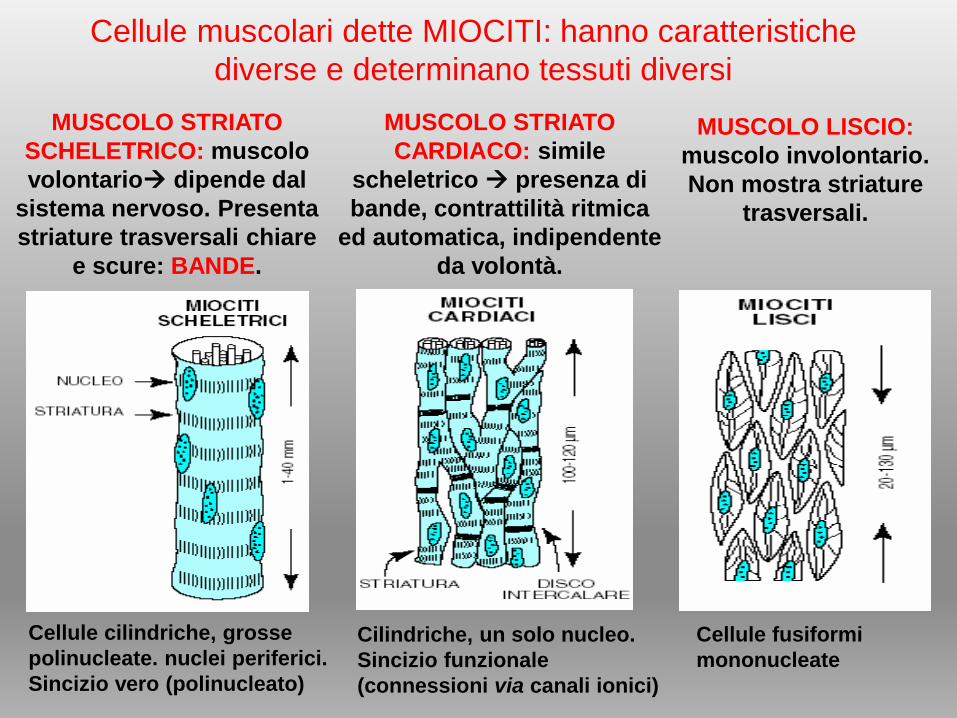
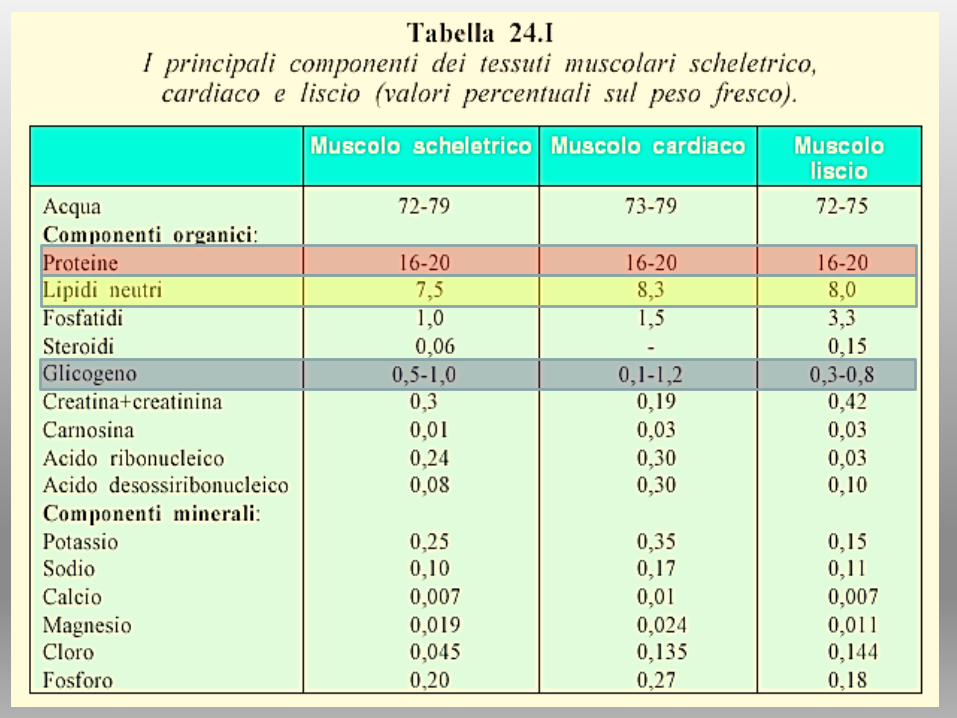
Cellule muscolari dette MIOCITI: hanno caratteristiche
diverse e determinano tessuti diversi
Cellule cilindriche, grosse
polinucleate. nuclei periferici.
Sincizio vero (polinucleato)
Cilindriche, un solo nucleo.
Sincizio funzionale
(connessioni via canali ionici)
Cellule fusiformi
mononucleate
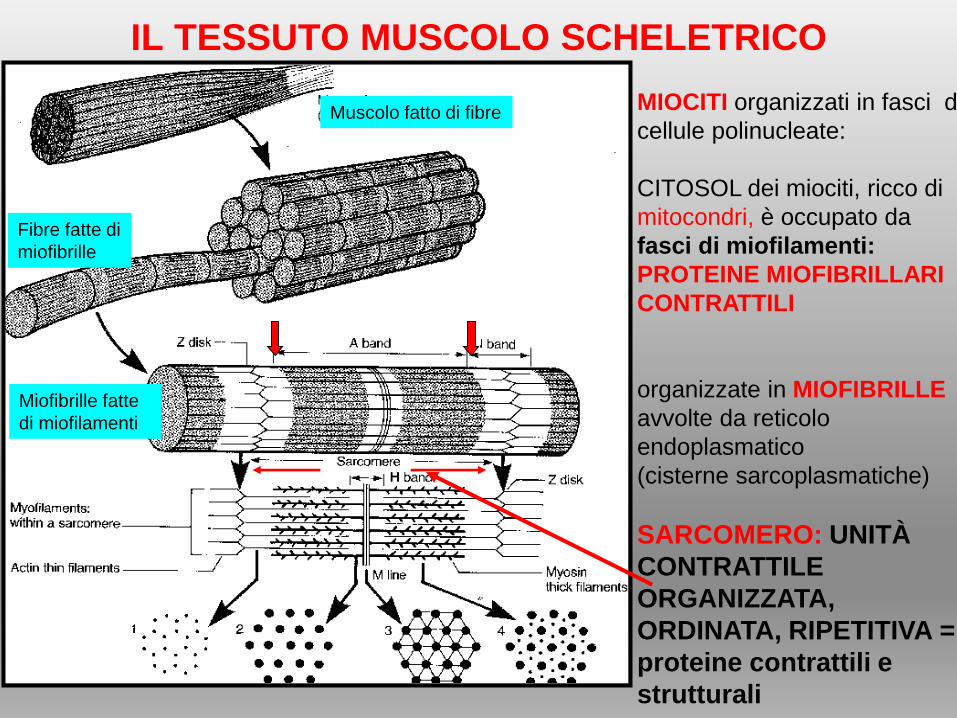
IL TESSUTO MUSCOLO SCHELETRICO
MIOCITI organizzati in fasci di
cellule polinucleate:
CITOSOL dei miociti, ricco di
mitocondri, è occupato da
fasci di miofilamenti:
PROTEINE MIOFIBRILLARI
CONTRATTILI
organizzate in MIOFIBRILLE
avvolte da reticolo
endoplasmatico
(cisterne sarcoplasmatiche)
SARCOMERO: UNITÀ
CONTRATTILE
ORGANIZZATA,
ORDINATA, RIPETITIVA =
proteine contrattili e
strutturali
Muscolo fatto di fibre
Fibre fatte di
miofibrille
Miofibrille fatte
di miofilamenti
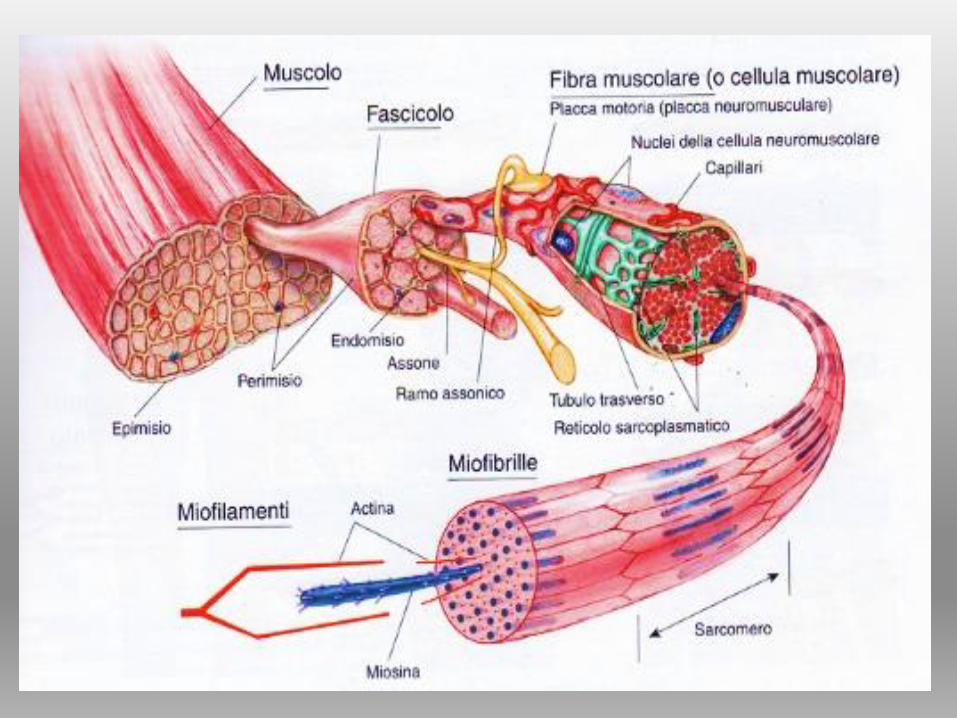
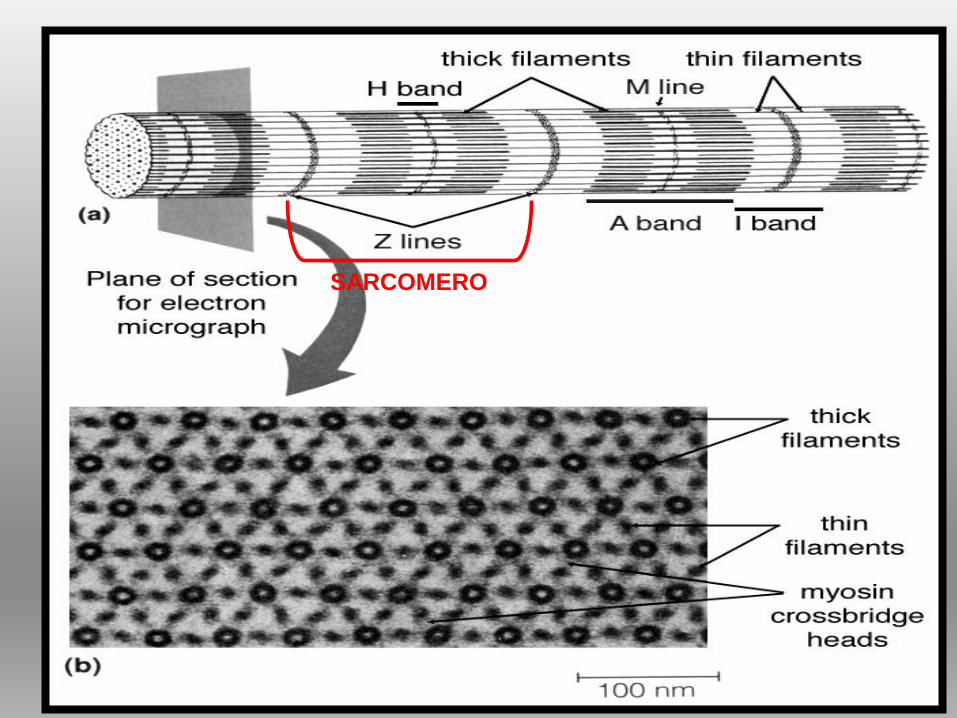
SARCOMERO
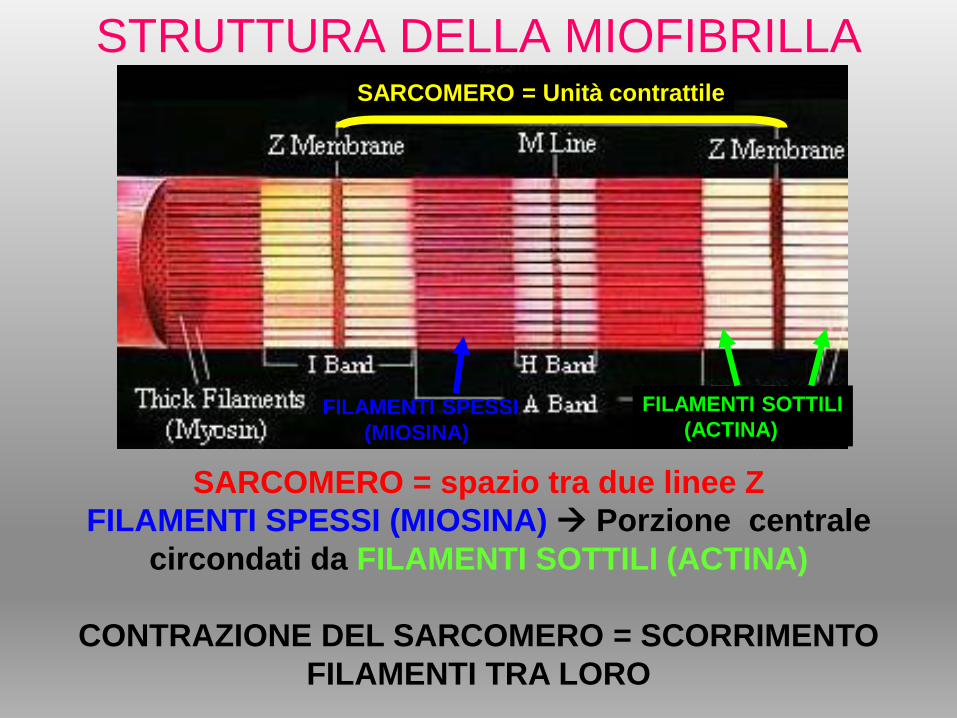
STRUTTURA DELLA MIOFIBRILLA
SARCOMERO = spazio tra due linee Z
FILAMENTI SPESSI (MIOSINA) Porzione centrale
circondati da FILAMENTI SOTTILI (ACTINA)
CONTRAZIONE DEL SARCOMERO = SCORRIMENTO
FILAMENTI TRA LORO
SARCOMERO = Unità contrattile
FILAMENTI SPESSI
(MIOSINA)
FILAMENTI SOTTILI
(ACTINA)
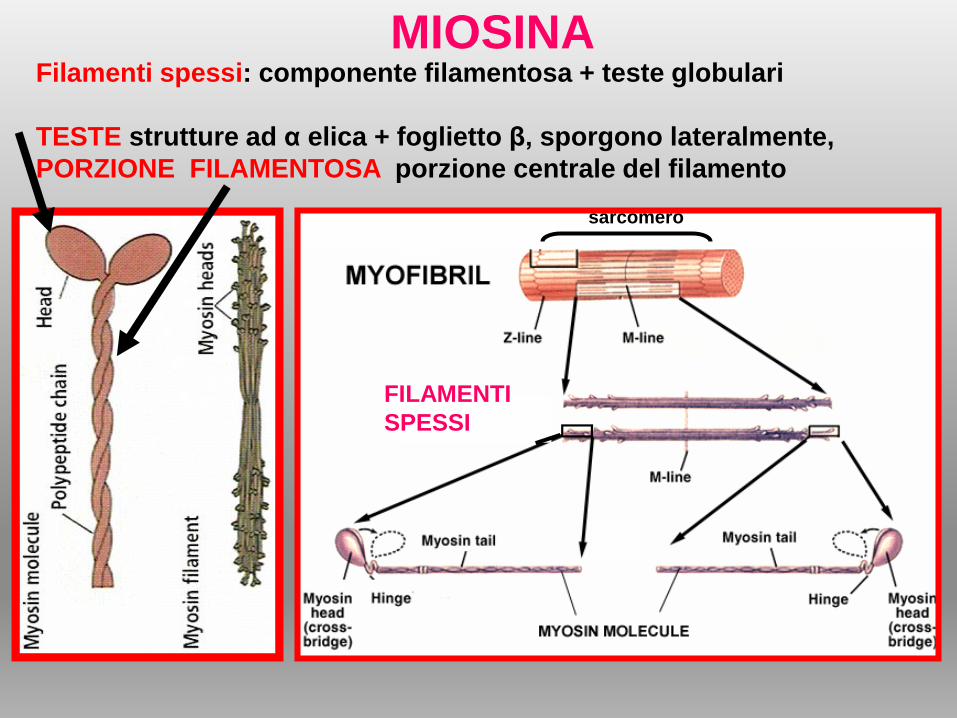
Filamenti spessi: componente filamentosa + teste globulari
TESTE strutture ad α elica + foglietto β, sporgono lateralmente,
PORZIONE FILAMENTOSA porzione centrale del filamento
MIOSINA
FILAMENTI
SPESSI
sarcomero
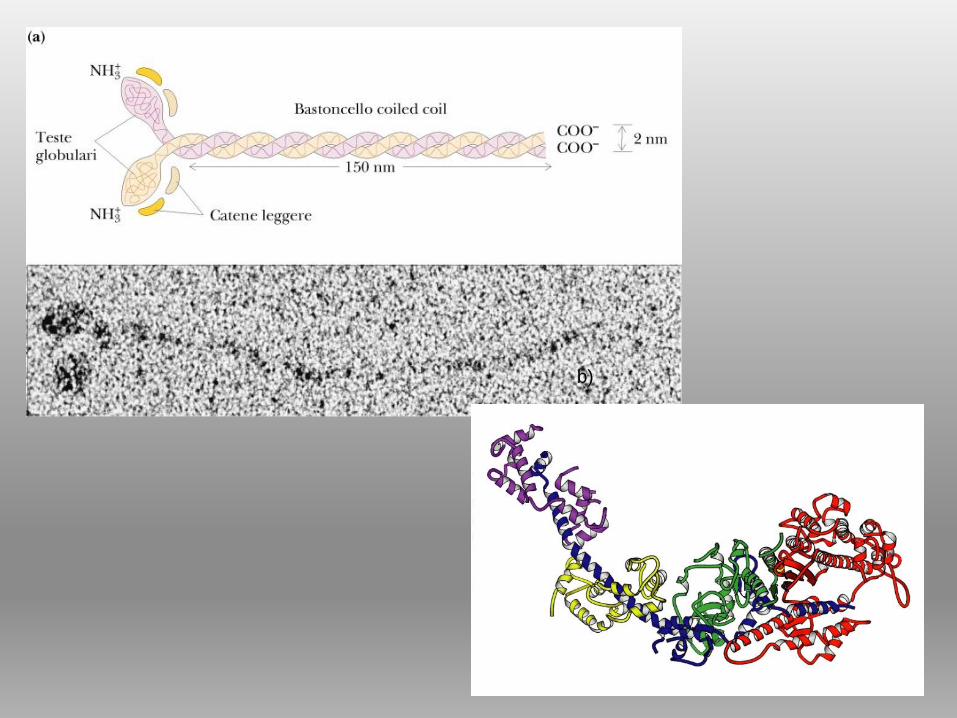
b)
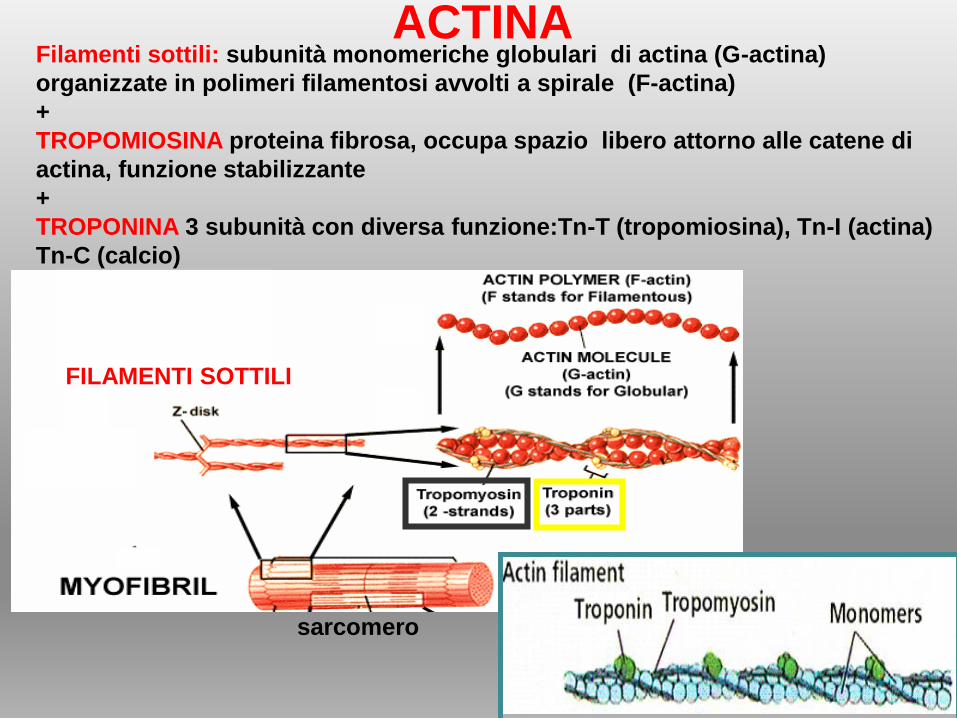
Filamenti sottili: subunità monomeriche globulari di actina (G-actina)
organizzate in polimeri filamentosi avvolti a spirale (F-actina)
+
TROPOMIOSINA proteina fibrosa, occupa spazio libero attorno alle catene di
actina, funzione stabilizzante
+
TROPONINA 3 subunità con diversa funzione:Tn-T (tropomiosina), Tn-I (actina)
Tn-C (calcio)
ACTINA
FILAMENTI SOTTILI
sarcomero
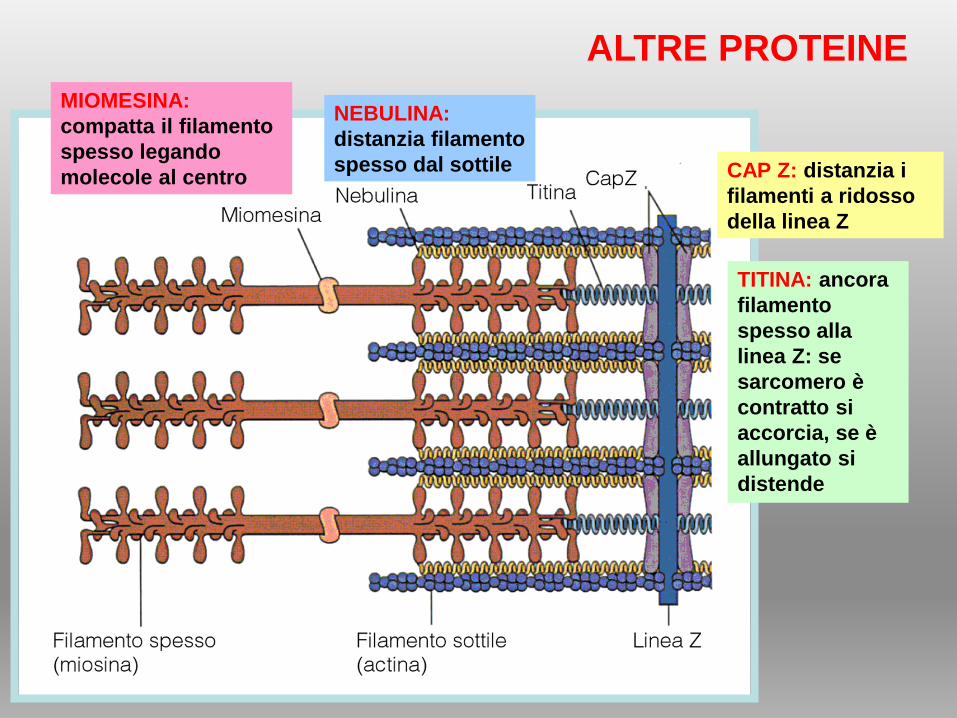
TITINA: ancora
filamento
spesso alla
linea Z: se
sarcomero è
contratto si
accorcia, se è
allungato si
distende
NEBULINA:
distanzia filamento
spesso dal sottile CAP Z: distanzia i
filamenti a ridosso
della linea Z
MIOMESINA:
compatta il filamento
spesso legando
molecole al centro
ALTRE PROTEINE
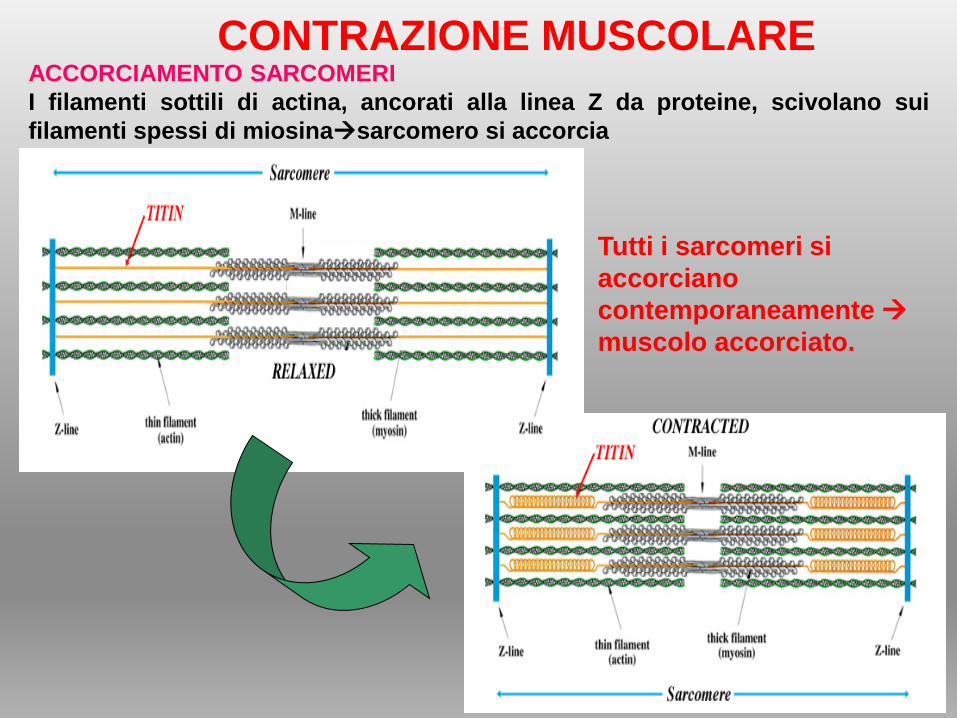
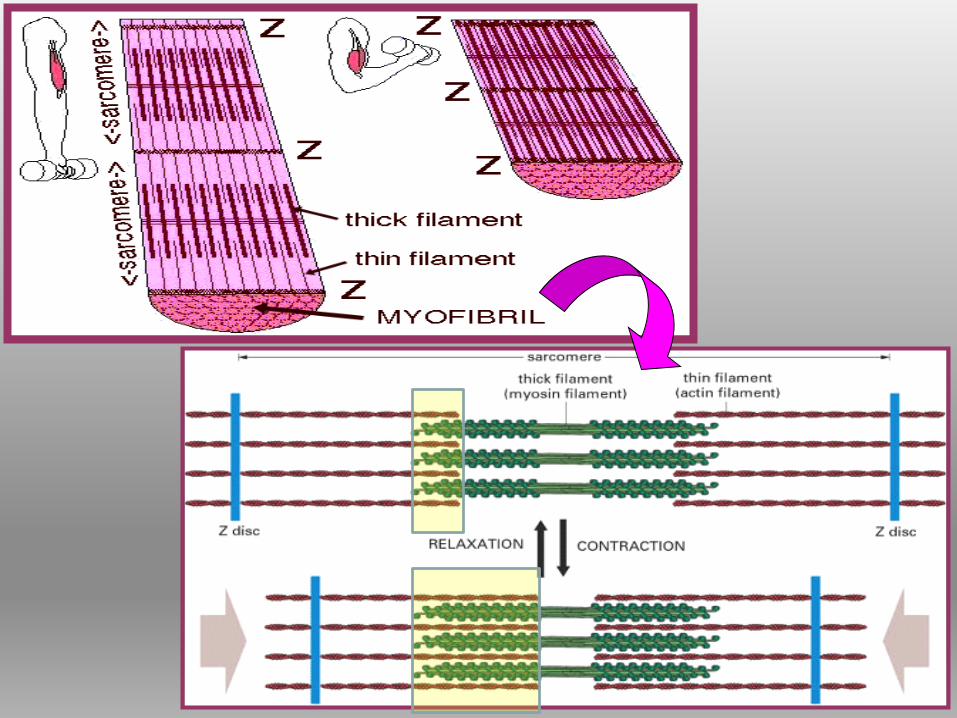
CONTRAZIONE MUSCOLAREACCORCIAMENTO SARCOMERI
I filamenti sottili di actina, ancorati alla linea Z da proteine, scivolano sui
filamenti spessi di miosinasarcomero si accorcia
Tutti i sarcomeri si
accorciano
contemporaneamente
muscolo accorciato.
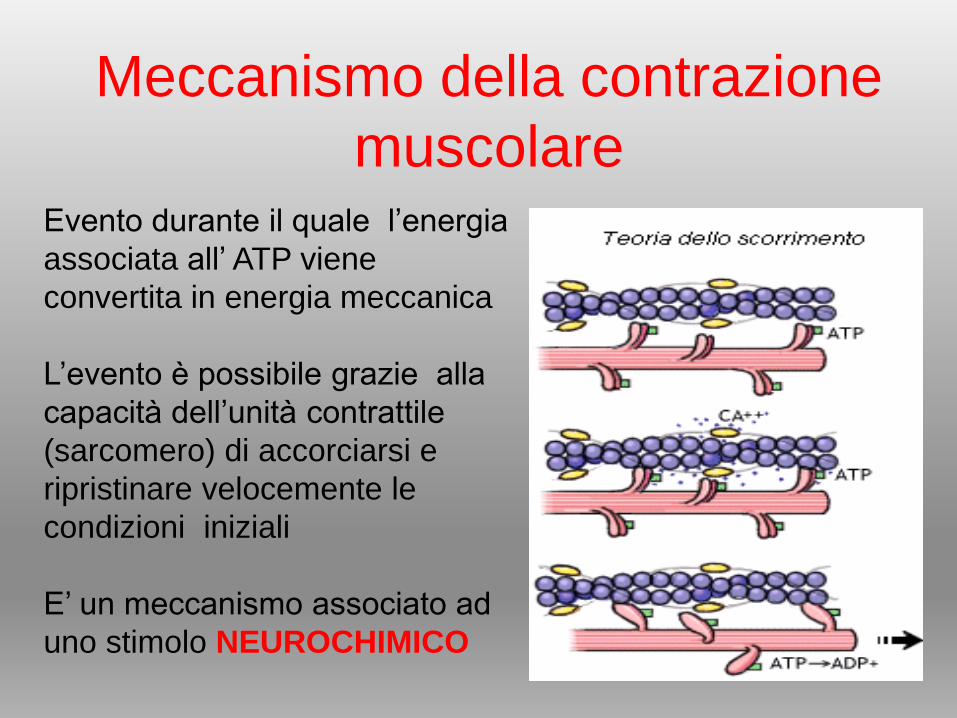
Meccanismo della contrazione
muscolareEvento durante il quale l’energia
associata all’ ATP viene
convertita in energia meccanica
L’evento è possibile grazie alla
capacità dell’unità contrattile
(sarcomero) di accorciarsi e
ripristinare velocemente le
condizioni iniziali
E’ un meccanismo associato ad
uno stimolo NEUROCHIMICO
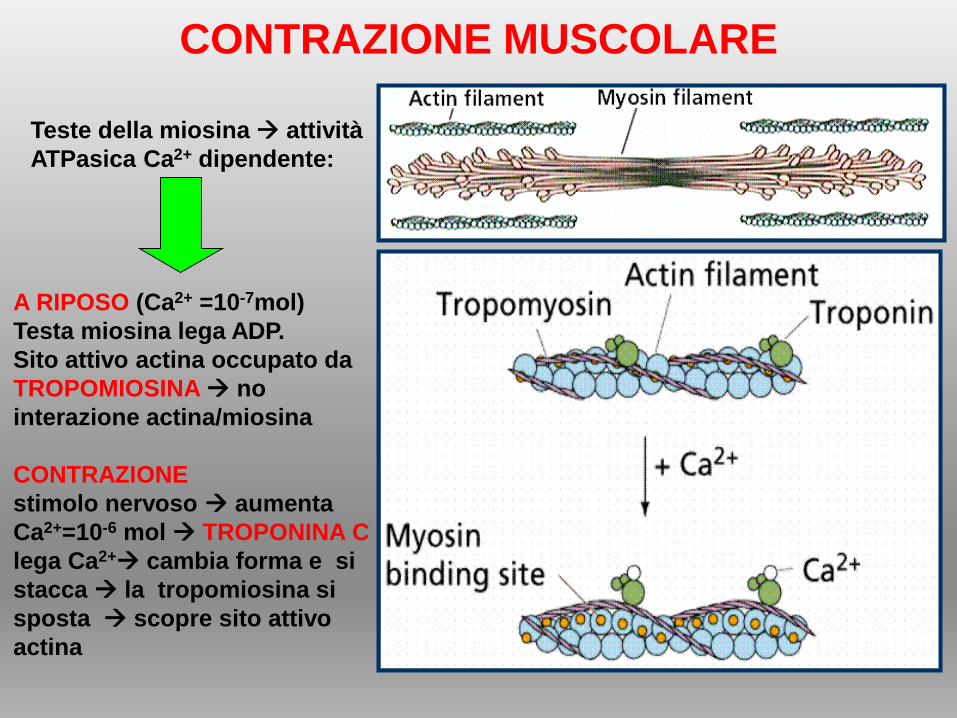
CONTRAZIONE MUSCOLARE
A RIPOSO (Ca2+ =10-7mol)
Testa miosina lega ADP.
Sito attivo actina occupato da
TROPOMIOSINA no
interazione actina/miosina
CONTRAZIONE
stimolo nervoso aumenta
Ca2+=10-6 mol TROPONINA C
lega Ca2+ cambia forma e si
stacca la tropomiosina si
sposta scopre sito attivo
actina
Teste della miosina attività
ATPasica Ca2+ dipendente:
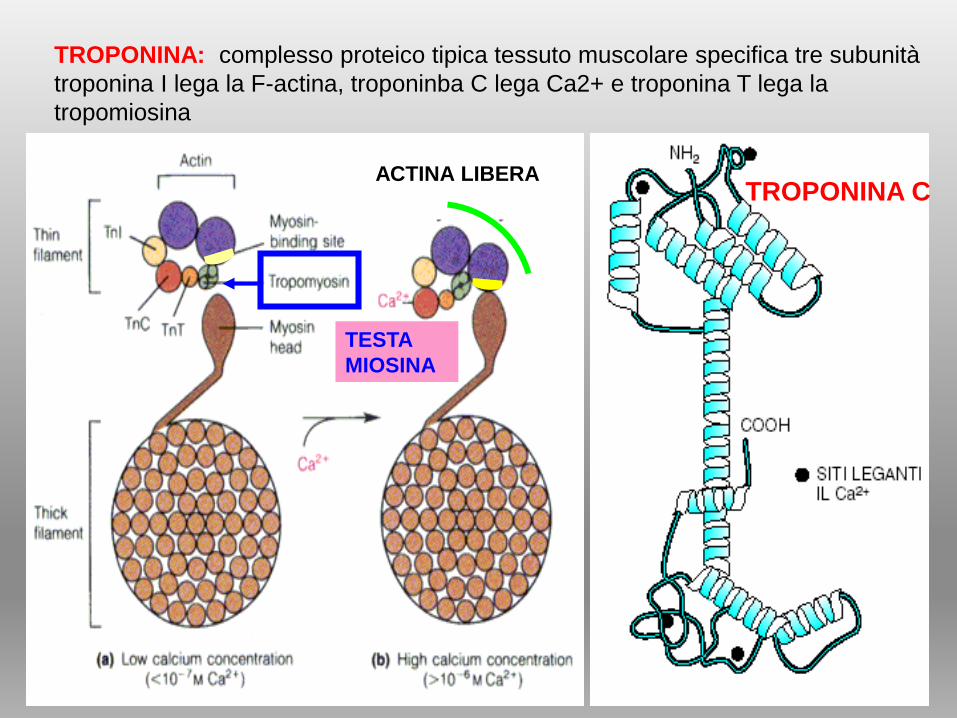
TROPONINA CACTINA LIBERA
TESTA
MIOSINA
TROPONINA: complesso proteico tipica tessuto muscolare specifica tre subunità
troponina I lega la F-actina, troponinba C lega Ca2+ e troponina T lega la
tropomiosina
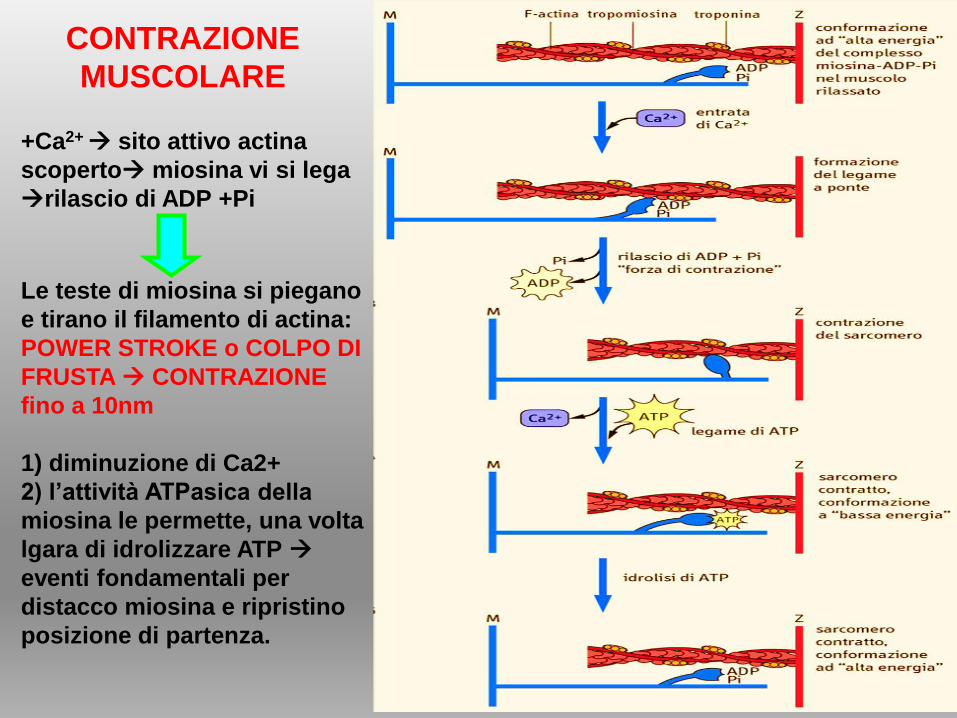
+Ca2+ sito attivo actina
scoperto miosina vi si lega
rilascio di ADP +Pi
Le teste di miosina si piegano
e tirano il filamento di actina:
POWER STROKE o COLPO DI
FRUSTA CONTRAZIONE
fino a 10nm
1) diminuzione di Ca2+
2) l’attività ATPasica della
miosina le permette, una volta
lgara di idrolizzare ATP
eventi fondamentali per
distacco miosina e ripristino
posizione di partenza.
CONTRAZIONE
MUSCOLARE
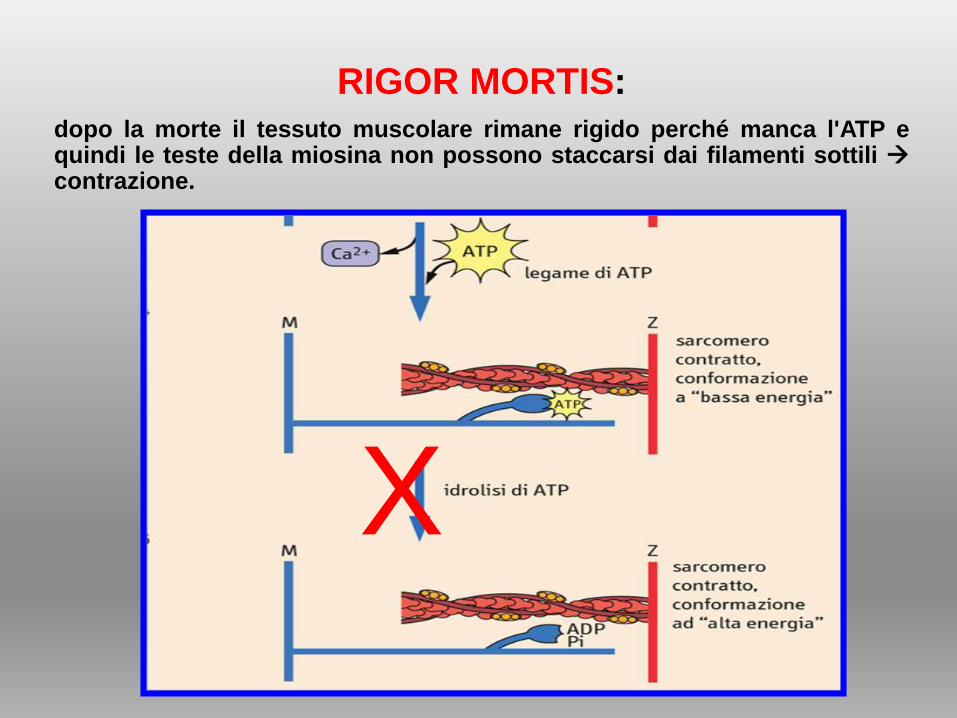
RIGOR MORTIS: dopo la morte il tessuto muscolare rimane rigido perché manca l'ATP equindi le teste della miosina non possono staccarsi dai filamenti sottili contrazione.
X
X
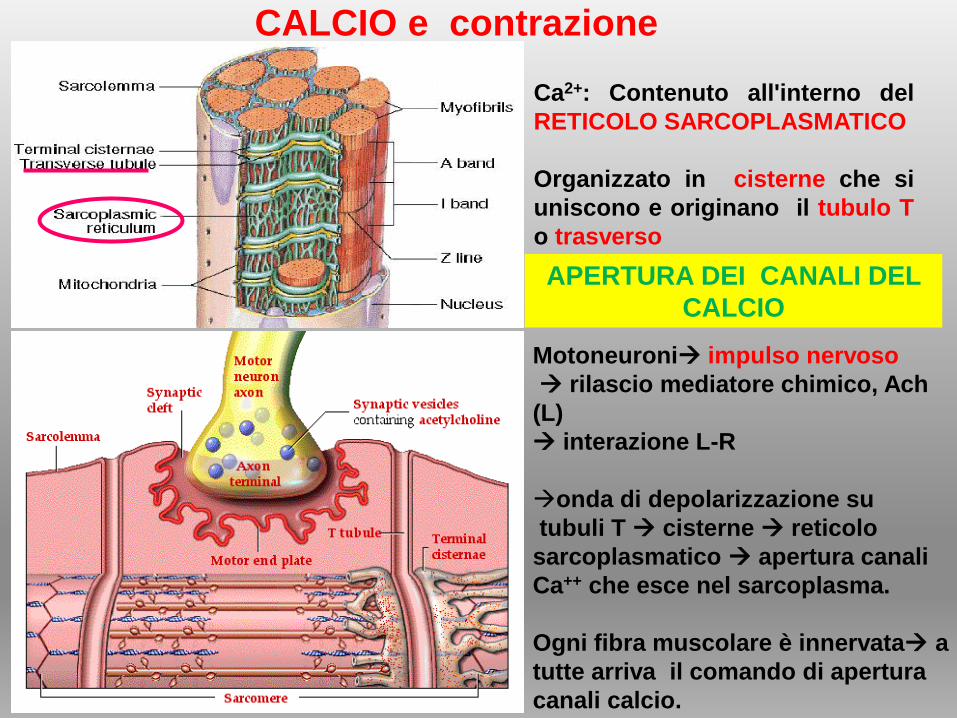
Ca2+: Contenuto all'interno del
RETICOLO SARCOPLASMATICO
Organizzato in cisterne che si
uniscono e originano il tubulo T
o trasverso
CALCIO e contrazione
Motoneuroni impulso nervoso
rilascio mediatore chimico, Ach
(L)
interazione L-R
onda di depolarizzazione su
tubuli T cisterne reticolo
sarcoplasmatico apertura canali
Ca++ che esce nel sarcoplasma.
Ogni fibra muscolare è innervata a
tutte arriva il comando di apertura
canali calcio.
APERTURA DEI CANALI DEL
CALCIO
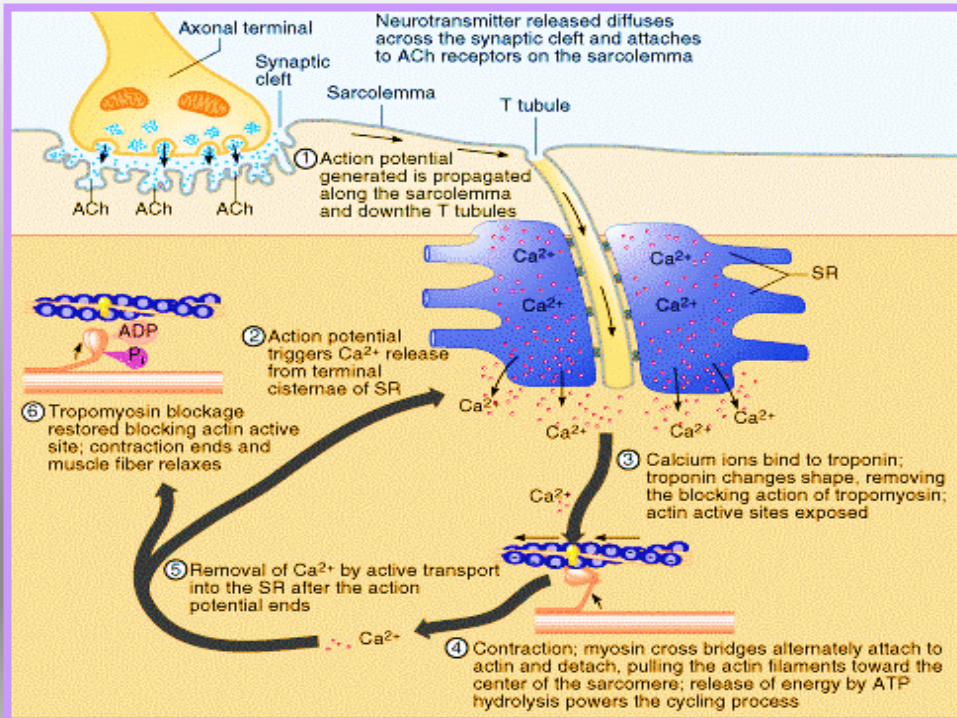
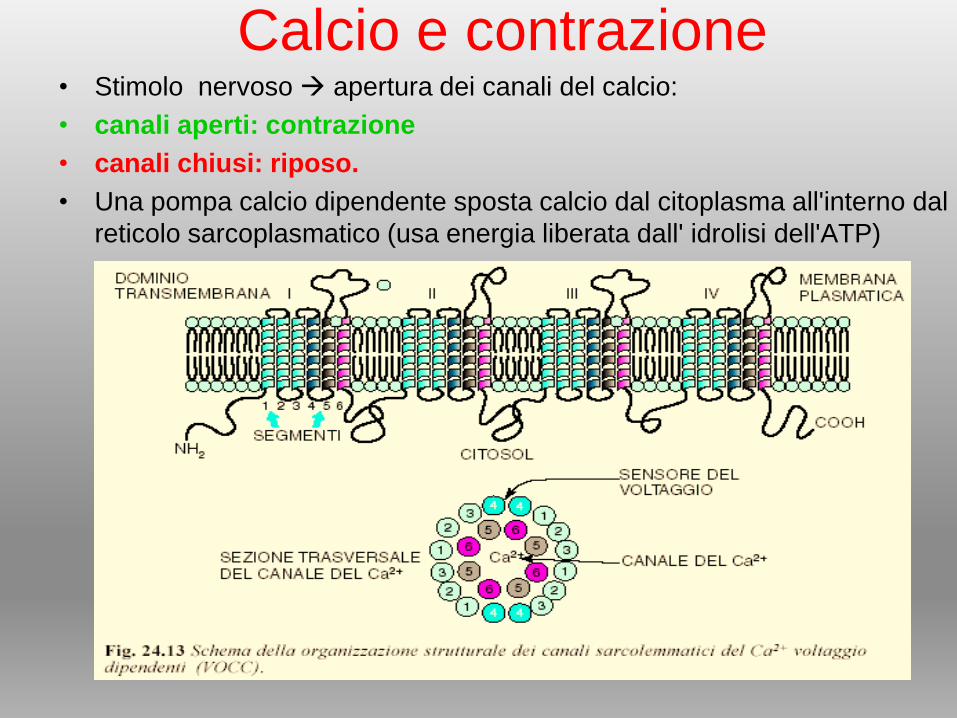
Calcio e contrazione• Stimolo nervoso apertura dei canali del calcio:
• canali aperti: contrazione
• canali chiusi: riposo.
• Una pompa calcio dipendente sposta calcio dal citoplasma all'interno dal
reticolo sarcoplasmatico (usa energia liberata dall' idrolisi dell'ATP)

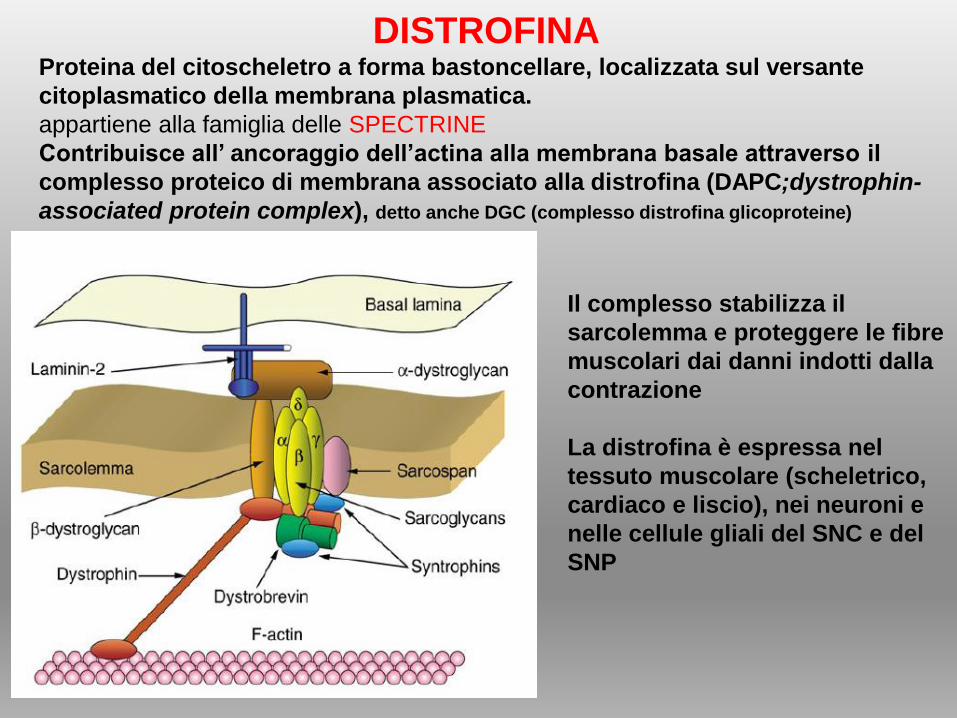
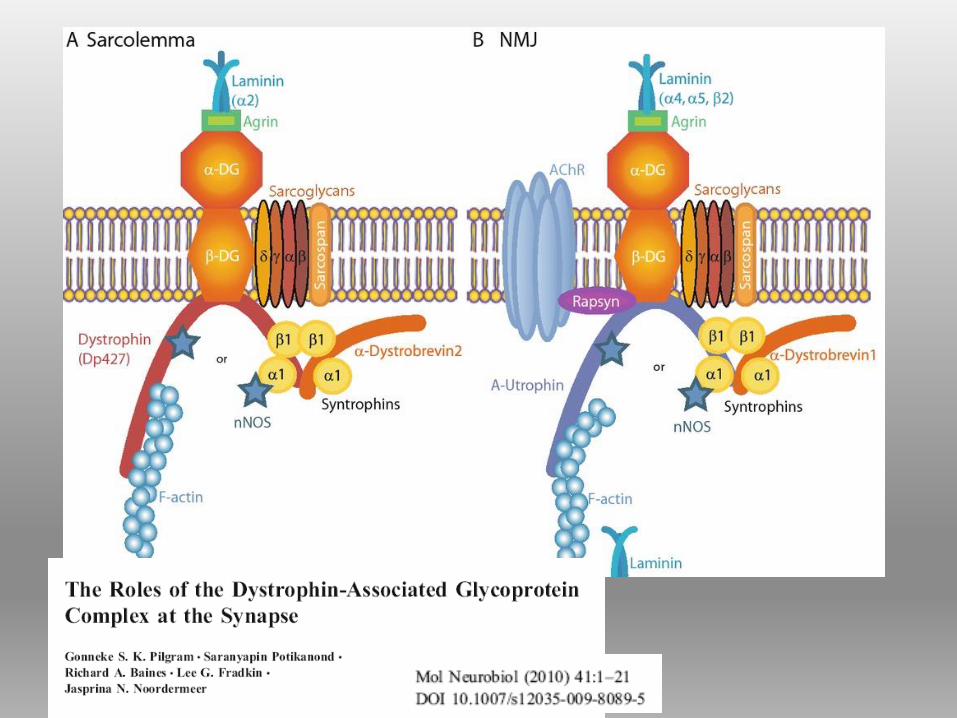
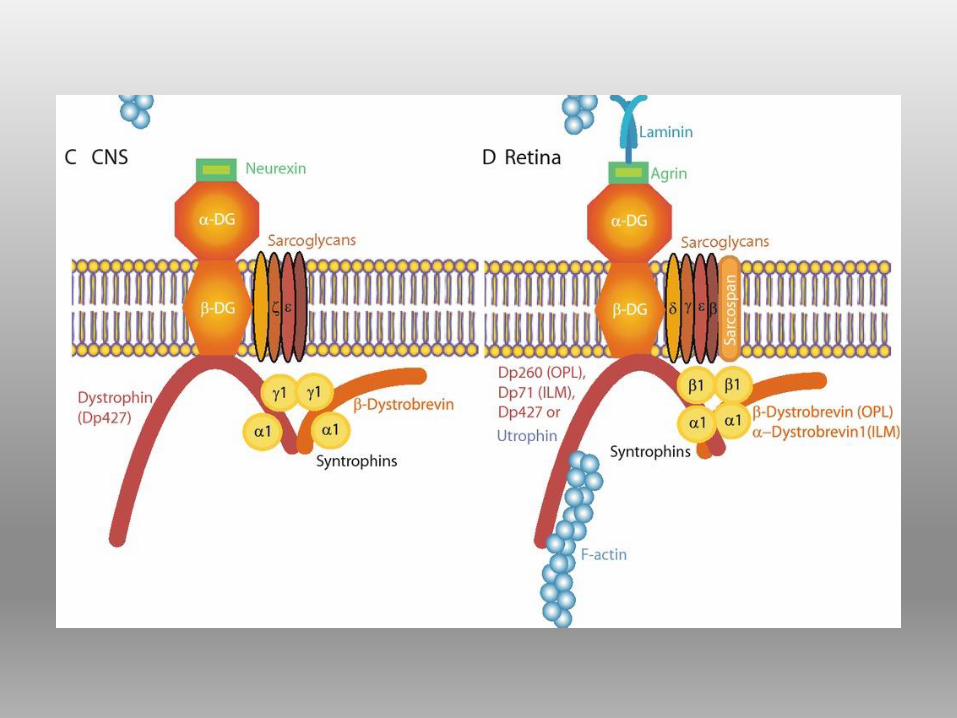
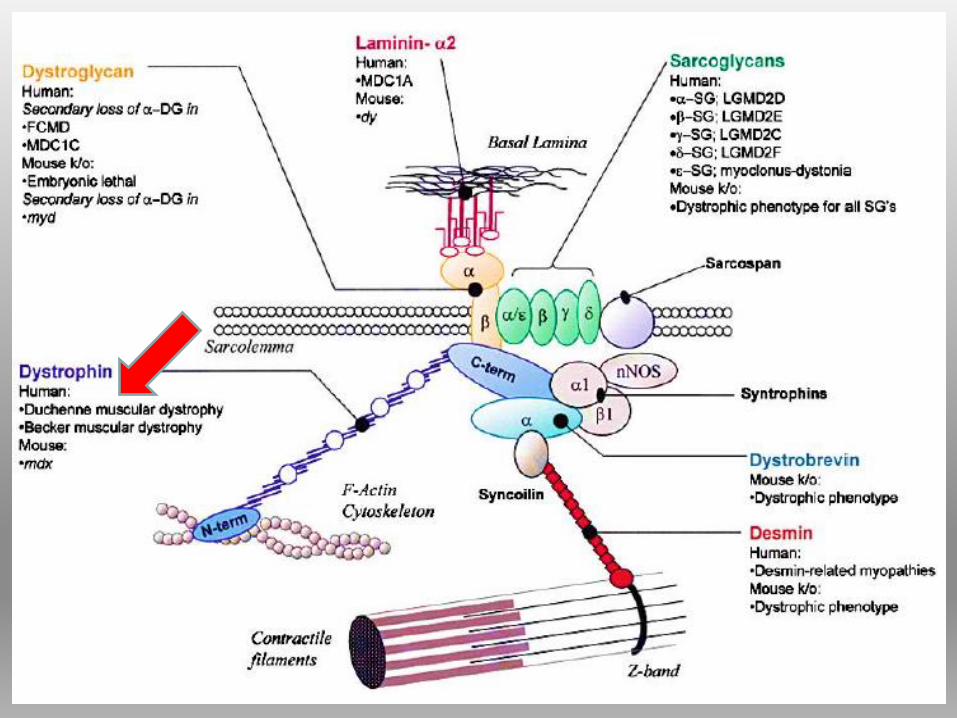
DISTROFINA Proteina del citoscheletro a forma bastoncellare, localizzata sul versante
citoplasmatico della membrana plasmatica.
appartiene alla famiglia delle SPECTRINE
Contribuisce all’ ancoraggio dell’actina alla membrana basale attraverso il
complesso proteico di membrana associato alla distrofina (DAPC;dystrophin-
associated protein complex), detto anche DGC (complesso distrofina glicoproteine)
Il complesso stabilizza il
sarcolemma e proteggere le fibre
muscolari dai danni indotti dalla
contrazione
La distrofina è espressa nel
tessuto muscolare (scheletrico,
cardiaco e liscio), nei neuroni e
nelle cellule gliali del SNC e del
SNP
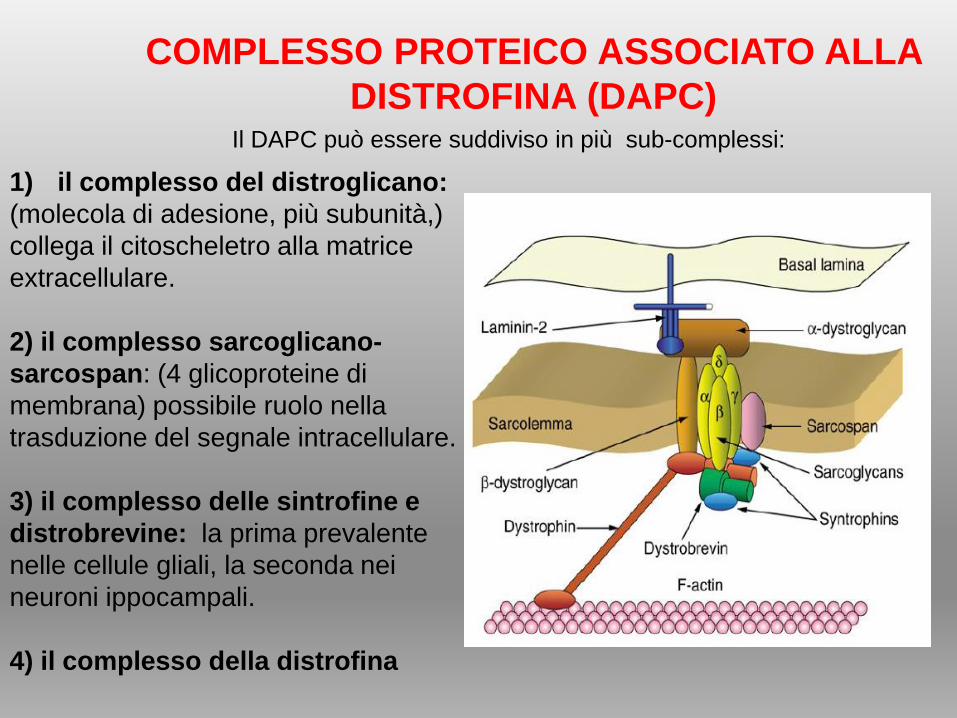
1) il complesso del distroglicano:
(molecola di adesione, più subunità,)
collega il citoscheletro alla matrice
extracellulare.
2) il complesso sarcoglicano-
sarcospan: (4 glicoproteine di
membrana) possibile ruolo nella
trasduzione del segnale intracellulare.
3) il complesso delle sintrofine e
distrobrevine: la prima prevalente
nelle cellule gliali, la seconda nei
neuroni ippocampali.
4) il complesso della distrofina
COMPLESSO PROTEICO ASSOCIATO ALLA
DISTROFINA (DAPC)Il DAPC può essere suddiviso in più sub-complessi:
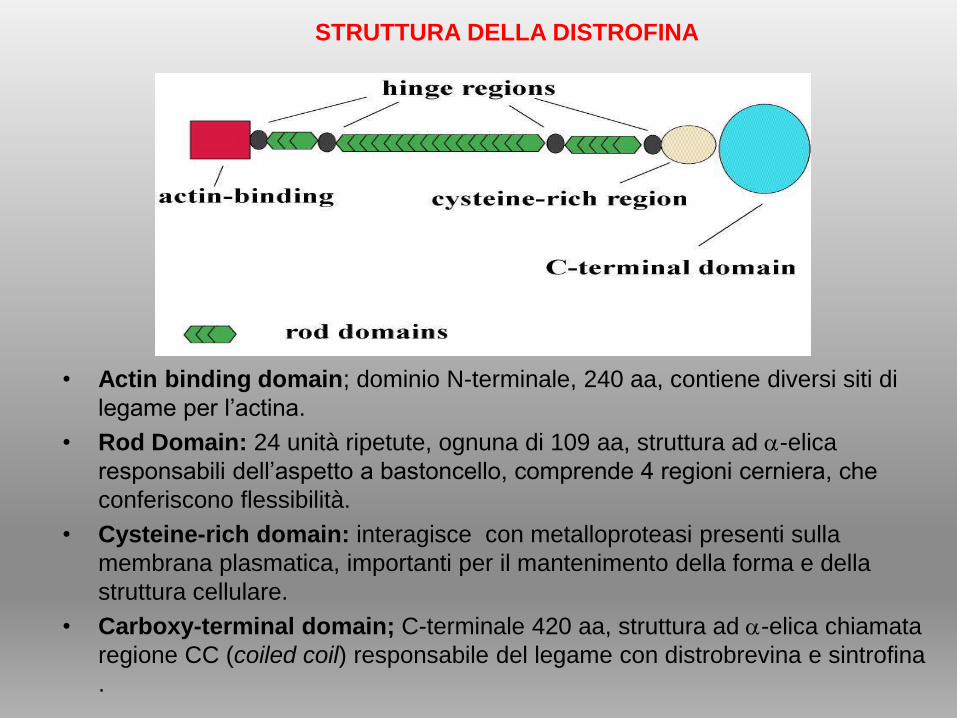
• Actin binding domain; dominio N-terminale, 240 aa, contiene diversi siti di
legame per l’actina.
• Rod Domain: 24 unità ripetute, ognuna di 109 aa, struttura ad -elica
responsabili dell’aspetto a bastoncello, comprende 4 regioni cerniera, che
conferiscono flessibilità.
• Cysteine-rich domain: interagisce con metalloproteasi presenti sulla
membrana plasmatica, importanti per il mantenimento della forma e della
struttura cellulare.
• Carboxy-terminal domain; C-terminale 420 aa, struttura ad -elica chiamata
regione CC (coiled coil) responsabile del legame con distrobrevina e sintrofina
.
STRUTTURA DELLA DISTROFINA
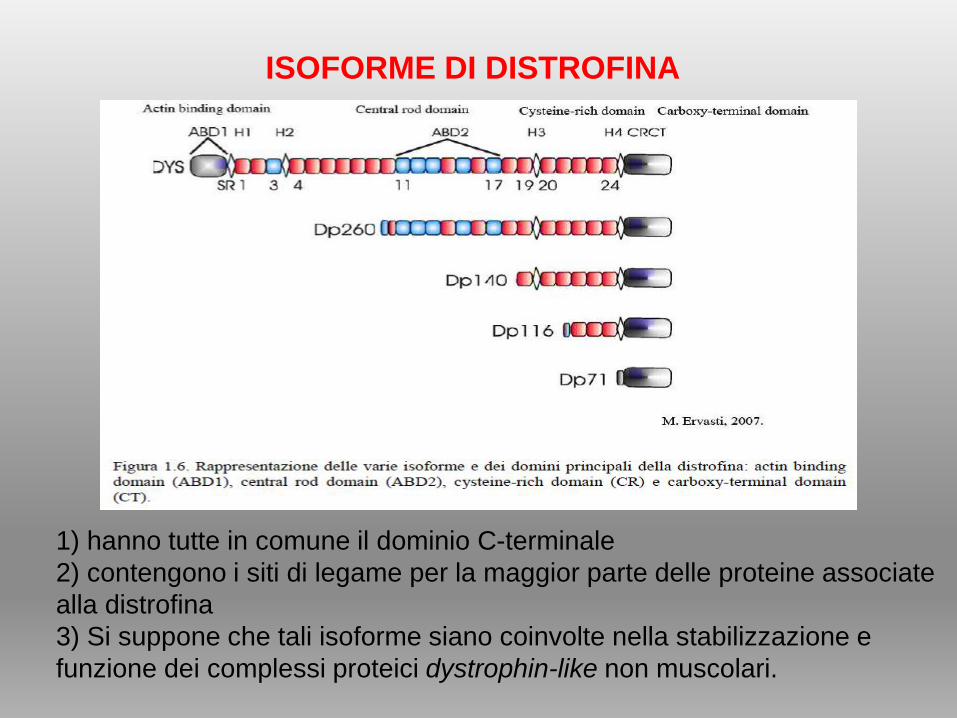
1) hanno tutte in comune il dominio C-terminale
2) contengono i siti di legame per la maggior parte delle proteine associate
alla distrofina
3) Si suppone che tali isoforme siano coinvolte nella stabilizzazione e
funzione dei complessi proteici dystrophin-like non muscolari.
ISOFORME DI DISTROFINA

• Ruolo strutturale:
La distrofina funge da ponte tra l’apparato contrattile intracellulare e la
matrice extra-cellulare, soprattutto a livello delle regioni miotendinee,
sottoposte a forte stress meccanico durante la contrazione muscolare.
In assenza di distrofina, diminuiscono anche tutte le proteine del
complesso; rendendo la membrana più suscettibile a rotture durante
l’attività contrattile.
• Ruolo nell’ aggregazione dei canali:
La distrofina organizza il citoscheletro, che a sua volta è responsabile
dell’aggregazione dei canali ionici e dei recettori per i neurotrasmettitori
RUOLO DELLA DISTROFINA
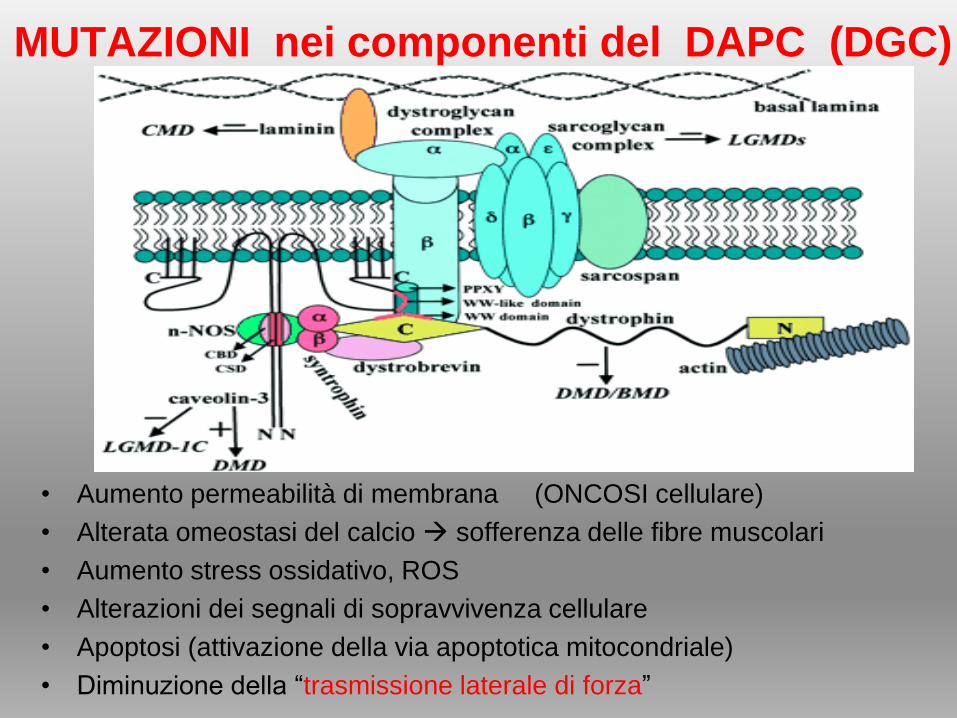
• Aumento permeabilità di membrana (ONCOSI cellulare)
• Alterata omeostasi del calcio sofferenza delle fibre muscolari
• Aumento stress ossidativo, ROS
• Alterazioni dei segnali di sopravvivenza cellulare
• Apoptosi (attivazione della via apoptotica mitocondriale)
• Diminuzione della “trasmissione laterale di forza”
MUTAZIONI nei componenti del DAPC (DGC)
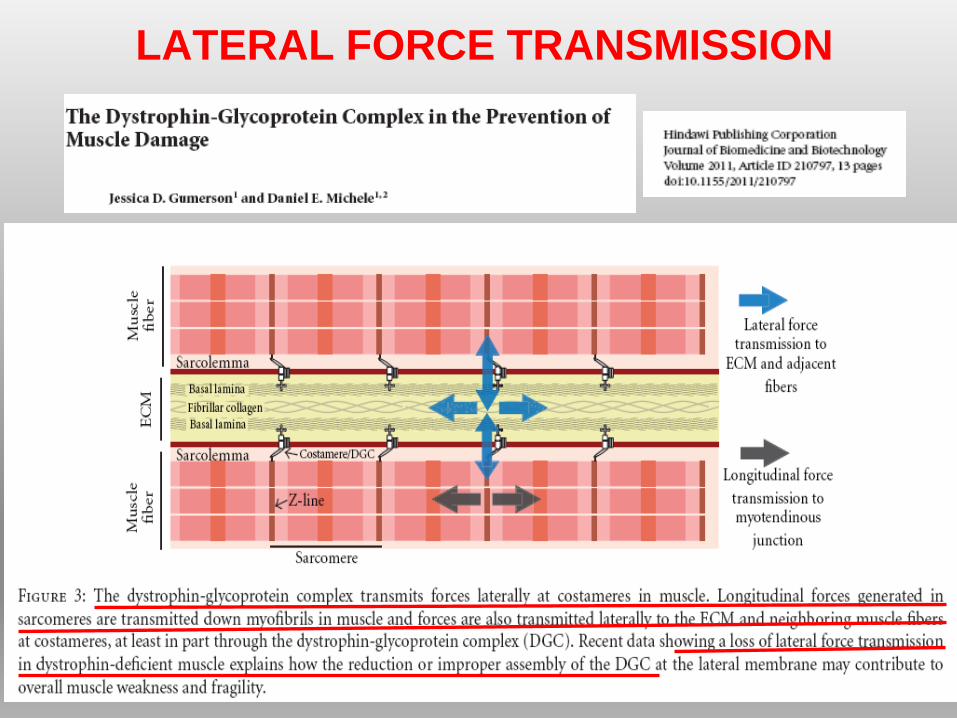
LATERAL FORCE TRANSMISSION
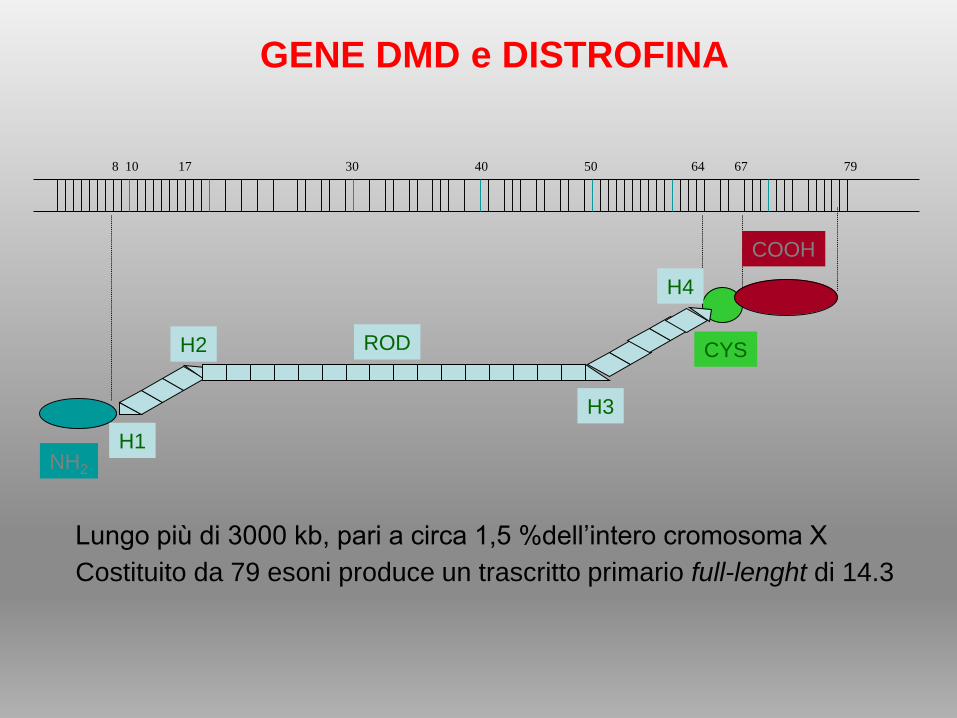
NH2
CYSROD
H1
H2
H4
H3
8 10 17 30 40 50 64 67 79
COOH
GENE DMD e DISTROFINA
Lungo più di 3000 kb, pari a circa 1,5 %dell’intero cromosoma X
Costituito da 79 esoni produce un trascritto primario full-lenght di 14.3

Almeno 7 trascritti diversi, generati da almeno 8 diversi promotori spesso
tessuto-specifici;
L’espressione del trascritto full-lenght è controllata da 3 promotori regolati in
modo indipendente per brain (B), muscle (M) e Purkinje (P).
Promotore B espressione primariamente nei neuroni corticali e nell’ippocampo,
Promotore P espresso nelle cellule cerebellari del Purkinje e muscolo
scheletrico.
Promotore M espresso ad alti livelli nel muscolo scheletrico, cardiomiociti , a
bassi livelli nelle cellule gliali nel cervello
Altri4 promotori interni portano alla produzione di un trascritto più corto,
codificante isoforme della distrofina tronche.
Tali promotori interni sono:
retinal (R), brain-3 (B3), Schwann cell (S), e general (G).
GENE DMD e DISTROFINA
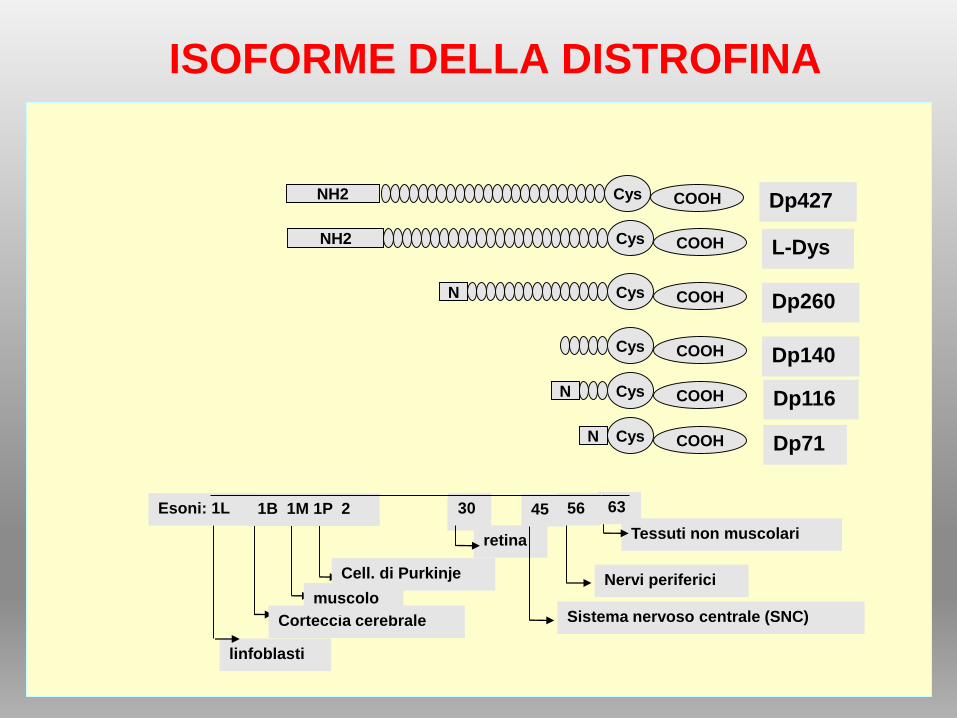
ISOFORME DELLA DISTROFINA
retina
Cys COOHNDp260
30
Dp140
Sistema nervoso centrale (SNC)
Cys COOH
45
Nervi periferici
Cys COOHN Dp116
56
Tessuti non muscolari
Cys COOHN Dp71
63
linfoblasti
Esoni: 1L
Cys COOHNH2L-Dys
Corteccia cerebrale
muscolo
Cell. di Purkinje
Cys COOHNH2 Dp427
1B 1M 1P 2

DISTROFIA di DUCHENNE• Prototipo delle distrofie muscolari. Tra le prime distrofie
muscolari descritte nel 1852.
• Più comune malattia fatale legata al cromosoma X.
• Incidenza: 1:3500 neonati maschi.
• Alterazione del gene per la distrofina: la distrofina è
assente
• Soggetti affetti: nel 65% dei casi si sono osservate
delezioni o duplicazioni.
• La mancanza totale di tale proteina provoca la morte
progressiva degli elementi muscolari.

Distrofina/DMD (Duchenne Muscular Dystrophy)
Il gene DMD è localizzato sulla banda Xp 21 del cromosoma X.
Le mutazioni sul gene sono.96% mutazioni frameshift, 30%
nuove mutazioni, 10-20% delle nuove avvengono nei gameociti

QUADRO CLINICO• Esordio tra i 3 e 5 anni. Disabilità entro i 10-12 anni
• Ingrossamento muscolare soprattutto dei muscoli del
polpaccio (PSEUDOIPERTROFIA).
• Deficit di forza più marcato nei DISTRETTI PROSSIMALI.
(Pelvi e cingolo scapolare).
• Difficoltà a stare seduto.
• Interessamento cardiaco (FIBROSI CARDIACA)
• Coinvolgimento muscoli lisci del tratto gastroenterico (vomito
episodico, dolore addominale e distensione gastrica)
• QI al di sotto della media normale +/- 75.
• La maggioranza dei pz muore di complicanze
dell’insufficienza respiratoria entro i 20 anni.
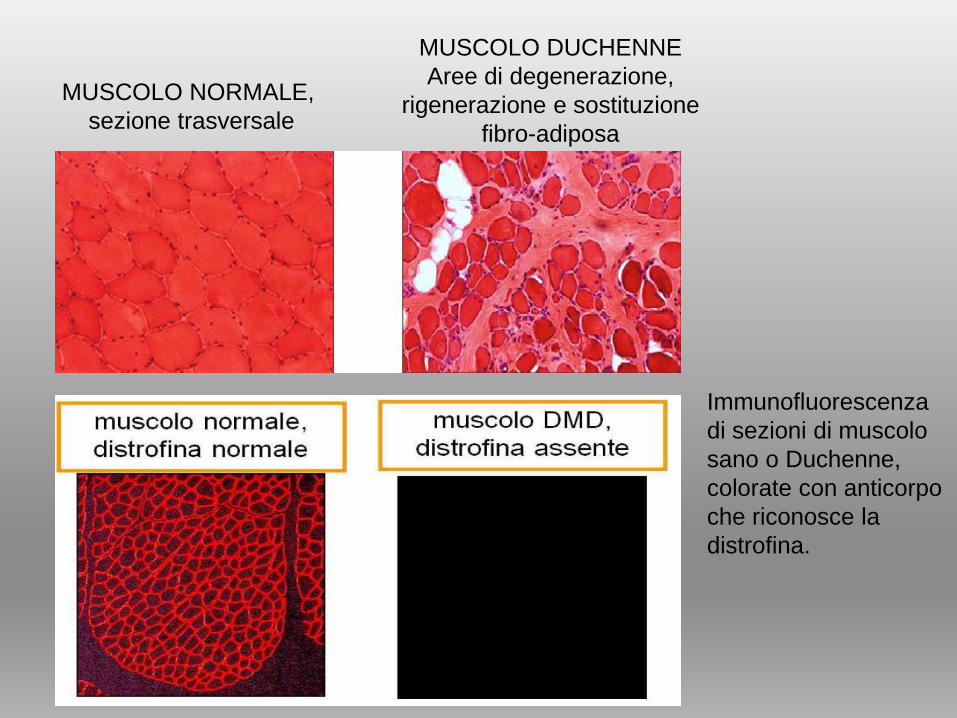
MUSCOLO NORMALE,
sezione trasversale
MUSCOLO DUCHENNE
Aree di degenerazione,
rigenerazione e sostituzione
fibro-adiposa
Immunofluorescenza
di sezioni di muscolo
sano o Duchenne,
colorate con anticorpo
che riconosce la
distrofina.
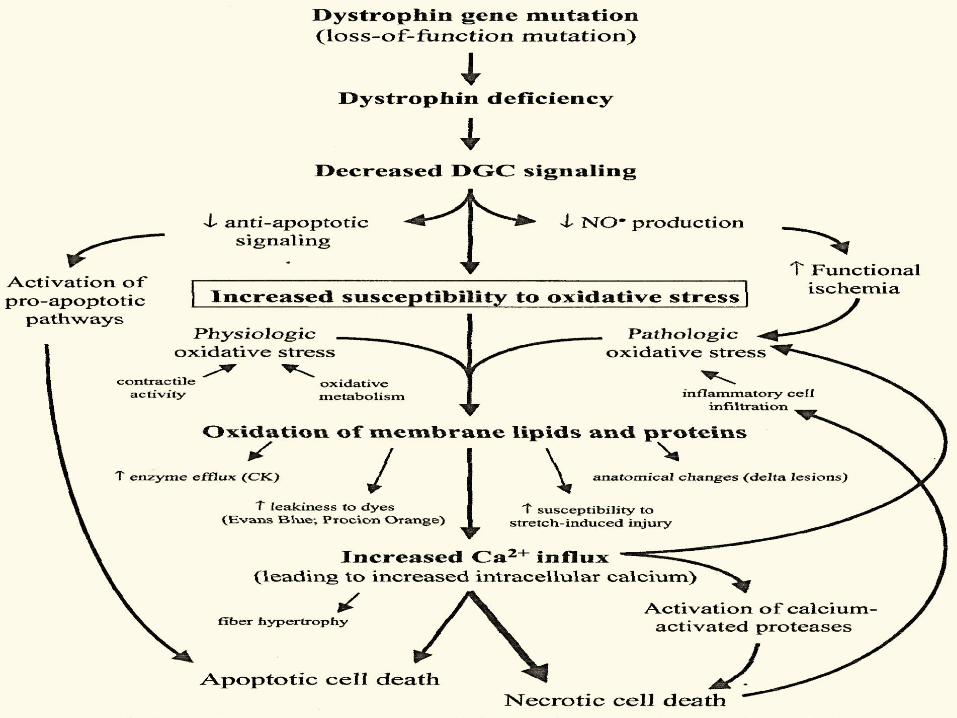
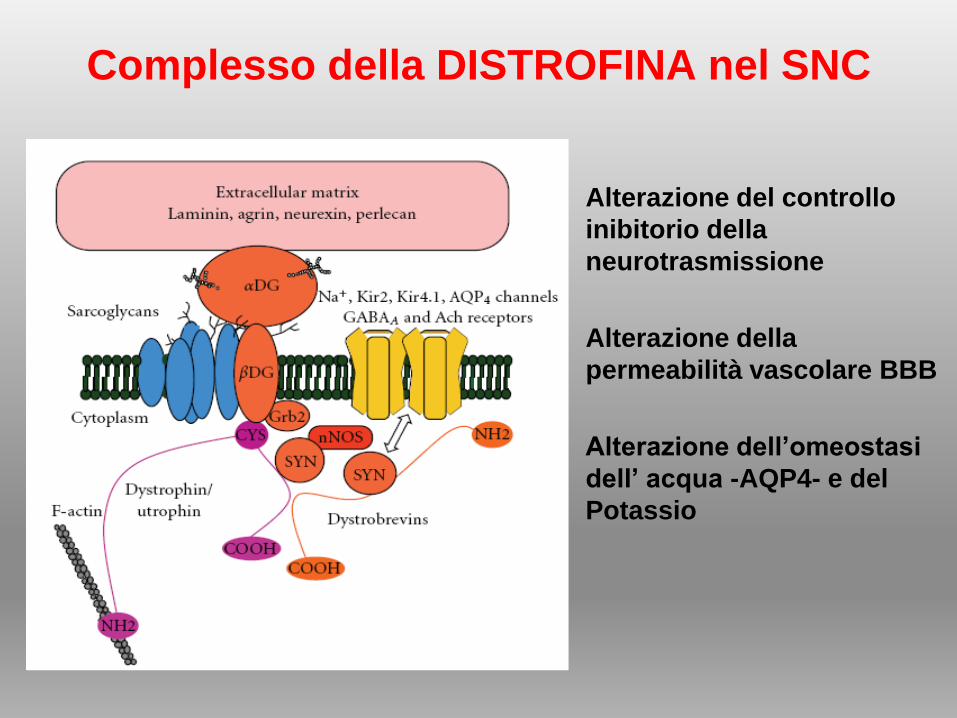
Complesso della DISTROFINA nel SNC
• Alterazione del controllo
inibitorio della
neurotrasmissione
• Alterazione della
permeabilità vascolare BBB
• Alterazione dell’omeostasi
dell’ acqua -AQP4- e del
Potassio