

Studio POSTER – Protocollo CONFIDENZIALE
POSTER
Prevalence Of familial hypercoleSTerolaemia (FH) in
ItalianPatients with coronary artERy disease
Numero di Protocollo K18
Versione 1.2 del 29 ottobre 2015
Promotori
Fondazione SISA (Società Italiana per lo studio dell’Arteriosclerosi)
Fondazione per il Tuo cuore HCF onlus

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 2/15
INDICE
1 INFORMAZIONI GENERALI ........................................................................................... 3
2 INFORMAZIONI AMMINISTRATIVE ............................................................................... 3
3 PAGINA DELLE FIRME PER CHAIRMAN E CO-CHAIRMAN DELLO STUDIO ............. 4
4 PAGINA DELLA FIRMA DELLO SPERIMENTATORE PRINCIPALE .............................. 5
5 LISTA DELLE ABBREVIAZIONI E DEGLI ACRONIMI .................................................... 6
6 INTRODUZIONE E RAZIONALE .................................................................................... 7
6.1 Fisiopatologia e diagnosi dell’ipercolesterolemia familiare ....................................... 7
6.2 Razionale dello studio .............................................................................................. 8
6.3 Diagnosi di ipercolesterolemia familiare in pazienti con cardiopatia ischemica ........ 9
7 OBIETTIVO DELLO STUDIO .......................................................................................... 9
8 POPOLAZIONE IN STUDIO ......................................................................................... 10
9 DISEGNO, METODOLOGIA E CONDUZIONE DELLO STUDIO .................................. 10
9.1 Disegno di Studio ....................................................................................................... 10
9.2 Durata dello Studio ..................................................................................................... 11
9.3 Metodologia ............................................................................................................... 11
9.3.1 Informazioni da raccogliere .............................................................................. 11
9.3.1.1 Dati per la diagnosi di ipercolesterolemia familiare: ................................... 11
9.3.2 Interruzione prematura dello studio.................................................................. 13
9.4 Gestione dei dati ........................................................................................................ 13
10 PUBBLICAZIONE DEI DATI ......................................................................................... 13
11 ACCESSO DIRETTO AI DATI ED AI DOCUMENTI ORIGINALI ................................... 13
12 ASPETTI ETICI ............................................................................................................. 13
13 REFERENZE BLIBLIOGRAFICHE ............................................................................... 14
APPENDICE 1. EAS/ESC GUIDELINES DIAGNOSTIC CRITERIA ..................................... 15

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 3/15
1 INFORMAZIONI GENERALI
PROTOCOLLO DI STUDIO
Titolo: Prevalence Of familial hypercoleSTerolaemia
(FH) in Italian Patients with coronary areERy disease
(POSTER)
Versione n.: 1.1 (Draft)
Data Versione: 28/10/2015
Titolo breve: POSTER Fase studio:
Studio osservazionale, multicentrico Numero di protocollo: K18
Promotore:
Fondazione per il Tuo cuore ONLUS - ANMCO
Fondazione SISA (Società Italiana per lo studio dell’Arteriosclerosi)
2 INFORMAZIONI AMMINISTRATIVE
Promotore
Fondazione per il Tuo cuore HCF onlus
Sede Legale Via La Marmora, 36 50121 Firenze
Sede Operativa Centro Studi ANMCO Via La Marmora, 34 50121 Firenze
Responsabile Operativo Dott. Aldo Pietro Maggioni
Legale Rappresentante Dott. Michele Massimo Gulizia
Fondazione SISA (Società Italiana per lo studio dell’Arteriosclerosi)
Sede Legale Via Balzaretti, 7 20133 Milano
Sede Operativa Via Balzaretti, 7 20133 Milano
Responsabile Operativo Prof. Alberico Luigi Catapano
Legale Rappresentante Prof. Maurizio Averna

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 4/15
3 PAGINA DELLE FIRME PER I CO-CHAIRMAN DELLO STUDIO
Studio POSTER
Approvato da:
Prof. Alberico Catapano 29.10.2015
(Co-Chairman) Firma Data
Dott. Michele Massimo Gulizia 29.10.2015
(Co-Chairman) Firma Data

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 5/15
4 PAGINA DELLA FIRMA DELLO SPERIMENTATORE PRINCIPALE
Studio POSTER
Ho letto questo protocollo e sono d'accordo a condurre questo studio in conformità con tutte le
disposizioni del protocollo e in accordo con la normativa vigente.
(Sperimentatore principale) Firma Data
Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 6/15
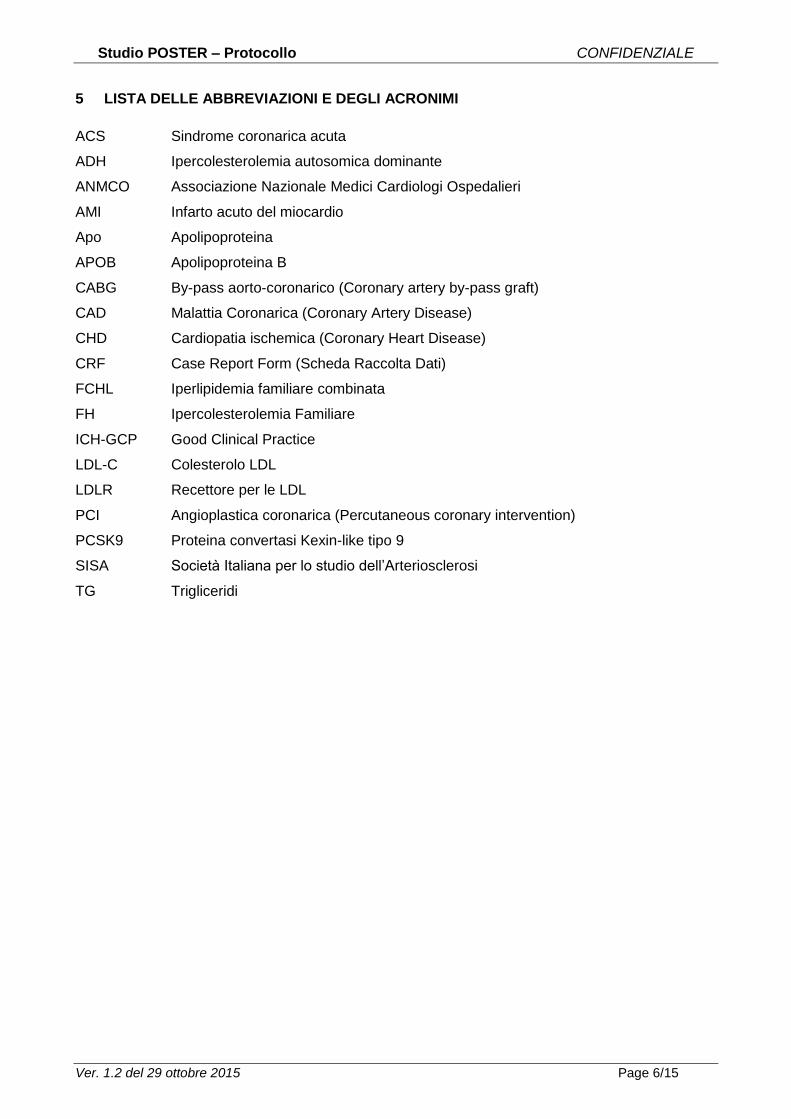
5 LISTA DELLE ABBREVIAZIONI E DEGLI ACRONIMI
ACS Sindrome coronarica acuta
ADH Ipercolesterolemia autosomica dominante
ANMCO Associazione Nazionale Medici Cardiologi Ospedalieri
AMI Infarto acuto del miocardio
Apo Apolipoproteina
APOB Apolipoproteina B
CABG By-pass aorto-coronarico (Coronary artery by-pass graft)
CAD Malattia Coronarica (Coronary Artery Disease)
CHD Cardiopatia ischemica (Coronary Heart Disease)
CRF Case Report Form (Scheda Raccolta Dati)
FCHL Iperlipidemia familiare combinata
FH Ipercolesterolemia Familiare
ICH-GCP Good Clinical Practice
LDL-C Colesterolo LDL
LDLR Recettore per le LDL
PCI Angioplastica coronarica (Percutaneous coronary intervention)
PCSK9 Proteina convertasi Kexin-like tipo 9
SISA Società Italiana per lo studio dell’Arteriosclerosi
TG Trigliceridi

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 7/15
6 INTRODUZIONE E RAZIONALE
6.1 Fisiopatologia e diagnosi dell’ipercolesterolemia familiare
L’ipercolesterolemia familiare (familial hypercholesterolaemia, FH) è una patologia che causa
l’esposizione a elevati livelli plasmatici di colesterolo LDL (LDL-C, low-density lipoprotein) durante
tutta la vita, aumentando quindi il rischio di malattia coronarica precoce (coronary artery disease,
CAD) (1). La FH è una malattia genetica co-dominante e può presentarsi in forma eterozigote ed
omozigote, con gravità diversa. Se non trattati i soggetti affetti da FH eterozigote, uomini e donne,
caratterizzati da livelli di colesterolo compresi tra 8-15 mmol/L (310-580 mg/dL), sviluppano
cardiopatia ischemica (Coronary heart disease, CHD) su base aterosclerotica rispettivamente
prima dei 55 e dei 60 anni, mentre i soggetti affetti da FH omozigote presentano livelli di
colesterolo di 12-30 mmol/L (460-1160 mg/dL) e sviluppano CHD in età giovanile, e, se non
sottoposti a terapia, muoiono prima dei 20 anni. La FH eterozigote, denominata anche
ipercolesterolemia autosomica dominante (ADH), è causata da mutazioni in eterozigosi nei geni
che codificano per proteine chiave coinvolte nel metabolismo del colesterolo LDL (2). Nello
specifico sono state ad ora identificate mutazioni con perdita di funzione nel gene codificante per il
recettore delle LDL (LDLR) (ADH-1), in quello codificante per il ligando del recettore
l’apolipoproteina B (APOB) (ADH-2) e nel gene codificante per la proteina PCSK9 (ADH-3).
Mutazioni causa di ADH-1 sono le più frequenti (3) e possono essere suddivise in 5 classi
funzionali che in vario modo riducono e/o alterano il ciclo funzionale del recettore: Classe 1,
mutazioni che determinano difetto di sintesi (allele nullo); Classe 2, mutazioni che determinano un
difetto di trasporto e maturazione del recettore dal reticolo endoplasmatico ruvido all’apparato del
Golgi; Classe 3, mutazioni che permettono la sintesi del recettore ed il suo trasferimento sulla
superficie cellulare ma comportano un difetto di legame del recettore con le LDL; Classe 4,
mutazioni che determinano una difettosa localizzazione del recettore nelle invaginazioni della
membrana cellulare rivestite dalla proteina clatrina e conseguentemente comportano un difetto di
internalizzazione del complesso LDL-LDLR; Classe 5, mutazioni che determinano un difetto di
riciclaggio del recettore per mancata dissociazione dal ligando (LDL) all’interno dell’endosoma e
mancato ritorno sulla superficie cellulare. Le mutazioni alla base dell’ADH-2 alterano invece la
capacità di legame dell’APOB con LDLR, mentre mutazioni con guadagno di funzione nel gene
PCSK9, una proteina coinvolta nella degradazione del LDLR sono alla base dell’ADH-3 (4,5).
Mutazioni in omozigosi dei tre geni sopra descritti o più spesso da mutazioni in eterozigosi
composta nei geni LDLR e nel gene codificante una proteina adattatrice (LDLRAP1), causano, in
loro assenza, una forma genetica di ipercolesterolemia a trasmissione recessiva (Autosomal
Recessive Hypercholesterolemia, ARH) (6). Rare sono anche le mutazioni in eterozigosi in due dei
quattro geni sopra descritti, che determinano un fenotipo intermedio tra quello degli FH eterozigoti
e degli FH omozigoti.
Da un punto di vista clinico l’identificazione dei soggetti affetti dalla patologia inizia con l’esclusione

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 8/15
delle forme secondarie, dipendenti dalla presenza di un’altra malattia primitiva che ha come
conseguenza un’alterazione del metabolismo lipidico (come l’ipotiroidismo o l’insufficienza renale)
o dall’utilizzo di farmaci o altre sostanze esogene che possono interferire sulle medesime vie
metaboliche. Successivamente, la diagnosi clinica si basa sui seguenti parametri: storia familiare e
anamnesi personale di CAD (usato CAD all’inizio) precoce, esame fisico per xantomi e arco
corneale, colesterolo LDL molto elevato a misurazioni ripetute, presenza di mutazioni causative.
Attualmente sulla base di questi parametri sono stati elaborati tre diversi criteri diagnostici: il
MedPed Program (Make early diagnosys, Prevent early dead) americano, i criteri del Simon
Broome Register Group nel Regno Unito e i criteri del Dutch Lipid Clinic Network (DLCN) elaborati
in Olanda. Il criterio MedPed è basato, sostanzialmente, sulla valutazione della colesterolemia in
rapporto all’età del soggetto. Per un adulto, la presenza di livelli di colesterolo totale superiori a
290 mg/dL è con buona approssimazione un valido criterio diagnostico. Gli altri criteri utilizzano un
sistema a punteggi basato sui livelli di colesterolo, sulla presenza di segni clinici (come xantomi
tendinei ed arco corneale) e sulla anamnesi familiare. Sono tre criteri validi e di relativa facile
applicazione nella pratica clinica, ma caratterizzati da bassa accuratezza. Alcune stime
suggeriscono infatti che i criteri clinici comportano il 25% di falsi negativi nei parenti di primo grado
di un probando. L’ indagine genetica, basata sul sequenziamento dei tre geni candidati, ha un
costo maggiore ma può permettere di arrivare a una diagnosi certa di ADH in circa il 60-80% dei
soggetti con FH probabile o certa ed alla caratterizzazione molecolare della patologia, facilitando
inoltre lo screening a cascata sui familiari del probando individuato.
6.2 Razionale dello studio
Non avendo una codifica indipendente nel sistema di classificazione internazionale delle malattie
definito dall’Organizzazione Mondiale della Sanita, risulta difficile fare una stima della prevalenza
di FH nella popolazione generale. Si ritiene generalmente che tra i soggetti di razza bianca vi sia
una prevalenza di FH eterozigote pari a 1/500 e di FH omozigote pari a 1/1.000.000 (1). Questa
prevalenza stimata teoricamente rappresenta però probabilmente una sottostima, in quanto si
basa su tassi di prevalenza in campioni di pazienti ricoverati in ospedale e su registri di patologia,
ed è influenzata dalla mortalità precoce dei pazienti con FH. Inoltre dati epidemiologici
suggeriscono che, anche basandosi su una prevalenza stimata a 1/500, la diagnosi venga
effettuata in meno dell’1% dei soggetti affetti nella maggioranza dei paesi, compresa l’Italia (7). In
un campione di 69016 soggetti della popolazione generale danese la prevalenza di individui
classificati con FH certa o FH probabile (secondo i criteri DLCN soggetti con punteggio >5 punti)
era pari a 0,73% (1/200), con FH possibile (criteri DLCN con punteggio 3-5 punti) 6,3% (1/16) e
con FH improbabile (Criteri DLCN <3 punti) 93% (8).
Utilizzando invece i criteri MEDPED la prevalenza di FH probabile era 0,80% (1/128), mentre la
prevalenza di FH certa o possibile secondo i criteri del Simon Broome Group era del 4,1% (1/25).
Una situazione analoga si può rinvenire nella popolazione italiana arruolata nello studio CHECK

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 9/15
(Cholesterol and Health: Evaluation, Control and Knowledge) dove su 6500 soggetti la percentuale
di soggetti con diagnosi di FH risulta essere di 1:250 (Catapano A.L. et al. Dati non pubblicati).
Nella stessa popolazione la prevalenza di CAD tra i soggetti con FH certa/probabile era del 33% e
solo il 48% di questi riceveva statine, mostrando un aumento del rischio di malattia coronarica di
circa 9 volte rispetto ad un aumento di 12 volte osservato in soggetti con FH certa/probabile non in
trattamento ipolipemizzante con statine (8). Questi dati mostrano chiaramente che nella
popolazione generale la FH è sotto-diagnosticata e che spesso i pazienti affetti da questa
patologia non ricevono un adeguato trattamento farmacologico.
6.3 Diagnosi di ipercolesterolemia familiare in pazienti con cardiopatia ischemica
La situazione sopra descritta riguardante la popolazione generale non migliora se si considerano
pazienti ricoverati per evento coronarico acuto (infarto del miocardio - AMI e/o angina instabile, -
ACS). I dati disponibili in letteratura al riguardo sono scarsi ma uno studio su 2153 pazienti con
AMI ricoverati in 20 ospedali nella regione dello Yorkshire ha mostrato come la prevalenza di FH
certa in questo campione, effettuata tramite diagnosi clinica, fosse del 12%, ma soprattutto che
solo nel 16% di questi soggetti la patologia era stata precedentemente diagnosticata e che solo
l’8% dei pazienti con FH erano in terapia con statine prima dell’evento acuto (9).
Considerando il carico cumulativo di colesterolo LDL a cui è sottoposto un soggetto affetto da FH,
la diagnosi ed il trattamento precoce rappresentano la modalità di intervento più efficace nel ridurre
la mortalità cardiovascolare in questi soggetti, per questo motivo implementare i metodi diagnostici
ed i trattamenti farmacologici sono obiettivi fondamentali per il miglioramento della gestione della
patologia (10, 11).
Il setting assistenziale offerto dai centri di cardiologia si presenta come momento di intercetto
ottimale per verificare la prevalenza di FH nei pazienti affetti da cardiopatia ischemica post-acuta
(AMI, ACS, intervento di rivascolarizzazione coronarica con bypass aorto-coronarico -CABG o
angioplastica coronarica percutanea - PCI). In questo contesto, in un’ottica di stratificazione
prognostica, può essere sistematicamente valutata la presenza di FH sia in un ambito di
prevenzione secondaria per pazienti identificati, che primaria nei familiari dei probandi individuati, e
implementare in essi percorsi diagnostici e terapeutici dedicati. Nel razionale dello studio non può
essere dimenticata la valenza di sensibilizzazione del mondo cardiologico al problema FH
arricchendone la formazione nell’affrontare adeguatamente una patologia così strettamente
collegata all’eziopatogenesi delle manifestazioni cliniche aterosclerotiche.
7 OBIETTIVO DELLO STUDIO
L’obiettivo primario del progetto è stabilire la prevalenza di ipercolesterolemia familiare (FH) in
pazienti con documentata malattia coronarica seguiti presso circa 100 strutture di cardiologia,
rappresentative in termini di distribuzione geografica del territorio italiano. L'analisi dei dati raccolti

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 10/15
permetterà anche di aumentare la sensibilizzazione dei medici cardiologi verso la patologia FH,
spesso sotto-diagnosticata. Obiettivo secondario dello studio sarà la validazione dei criteri
diagnostici del Dutch Lipid Clinic Network (DLCN), riportati in Appendice 1, nella popolazione
italiana affetta da malattia coronarica (12).
La caratterizzazione dei pazienti effettuata nello studio consentirà inoltre di derivare priorità per
intraprendere interventi sanitari mirati e per migliorare la diagnosi di FH nella popolazione generale
tramite lo screening a cascata dei familiari dei probandi con diagnosi di ipercolesterolemia familiare
certa, permettendo così l’individuazione precoce di pazienti a rischio cardiovascolare molto alto.
8 POPOLAZIONE IN STUDIO
In questo studio saranno reclutati 6000 pazienti adulti (età > 18 anni) di qualunque sesso, seguiti
presso 100 centri italiani di cardiologia.
I pazienti reclutati dovranno avere una documentata malattia coronarica, più precisamente:
sindrome coronarica acuta
Intervento di angioplastica coronarica (PCI)
Intervento di by-pass aorto-coronarico (CABG)
L’evento indice, tra quelli sopra descritti, dovrà essere occorso e documentato nell’intervallo tra 15
giorni e 8 settimane precedenti l’arruolamento. I pazienti reclutati nello studio saranno in grado di
comprenderne le procedure e di accettare volontariamente di partecipare alla ricerca fornendo il
consenso informato scritto. Saranno esclusi dallo studio i pazienti non in grado di comprendere le
procedure dello studio e di fornire volontariamente il consenso informato scritto.
Al termine dello studio ai pazienti con diagnosi di FH probabile o sicura verrà raccomandato di
continuare la gestione di questa patologia presso uno dei circa 40 centri, esistenti sul territorio
nazionale, specializzati nella cura delle dislipidemie appartenenti al network LIPIGEN (LIPid
Transport disorders Italian GEnetic Network).
9 DISEGNO, METODOLOGIA E CONDUZIONE DELLO STUDIO
9.1 Disegno di Studio
Lo studio POSTER è uno studio di natura osservazionale, multicentrico, prospettico, volto
all’individuazione di pazienti affetti da ipercolesterolemia familiare tra i pazienti con documentata
malattia coronarica.
I pazienti partecipanti allo studio non verranno sottoposti ad alcuna procedura che esuli dalla
normale pratica clinica quotidiana; allo stesso modo, le variabili cliniche che verranno raccolte per
lo studio sono quelle che vengono comunemente raccolte dal Medico nella pratica clinica

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 11/15
quotidiana.
9.2 Durata dello Studio
Lo studio in oggetto si propone essere uno studio osservazionale prospettico con reclutamento di
6000 pazienti, con un periodo di arruolamento previsto di 12 mesi. In ogni centro partecipante i 12
mesi decorreranno dalla data di inclusione del primo paziente.
9.3 Metodologia
Lo studio è a carattere osservazionale, non interventistico.
I pazienti saranno trattati secondo quanto previsto dalla pratica clinica in accordo al giudizio del
Medico. Rientra, in particolare, nella pratica clinica corrente di pazienti affetti da malattia
coronarica, l’anamnesi della storia familiare e personale di malattia cardiovascolare precoce e di
dislipidemia e l’analisi biochimica del profilo (12) lipidico.
I livelli ematici dei lipidi per la diagnosi clinica di FH dovranno essere valutati almeno 15 giorni
dopo l’evento indice, momento in cui si è ragionevolmente sicuri che l’effetto della terapia
ipolipemizzante eventualmente in atto sia massimo. I valori ottenuti nei pazienti in terapia
ipolipemizzante, verranno aggiustati sulla base della percentuale di decremento riconosciuta dalla
letteratura alle singole molecole e dei singoli dosaggi (definizione di bassa dose di statina:
Atorvastatina ≤10 mg, Fluvastatina ≤40 mg, Lovastatina ≤20 mg, Pravastatina ≤20 mg,
Rosuvastatina ≤5 mg, Simvastatina <20 mg).
Il medico sperimentatore dovrà verificare ed identificare presso il proprio centro gli eventuali
soggetti eleggibili da poter includere nello studio.
Un soggetto sarà considerato arruolato nello studio solo dopo che il medico sperimentatore avrà
ottenuto il consenso informato scritto da parte del soggetto stesso.
9.3.1 Informazioni da raccogliere
Per ogni soggetto arruolato nello studio, lo sperimentatore dovrà registrare in CRF le informazioni
disponibili, di seguito riportate, oltre alla disponibilità del consenso informato scritto, datato e
firmato:
9.3.1.1 Dati per la diagnosi di ipercolesterolemia familiare:
Dati demografici: data di nascita, sesso, origine geografica e appartenenza etnica
Anamnesi e dati antropometrici: altezza, peso, fumo.
Esame obiettivo
Anamnesi familiare
Storia Clinica di patologia cardiovascolare precoce e dislipidemia
Patologie concomitanti
Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 12/15
Terapie concomitanti
Esami biochimici (LDL-C, HDL-C, TG, glucosio, creatinina)
Diagnosi molecolare di ipercolesterolemia familiare
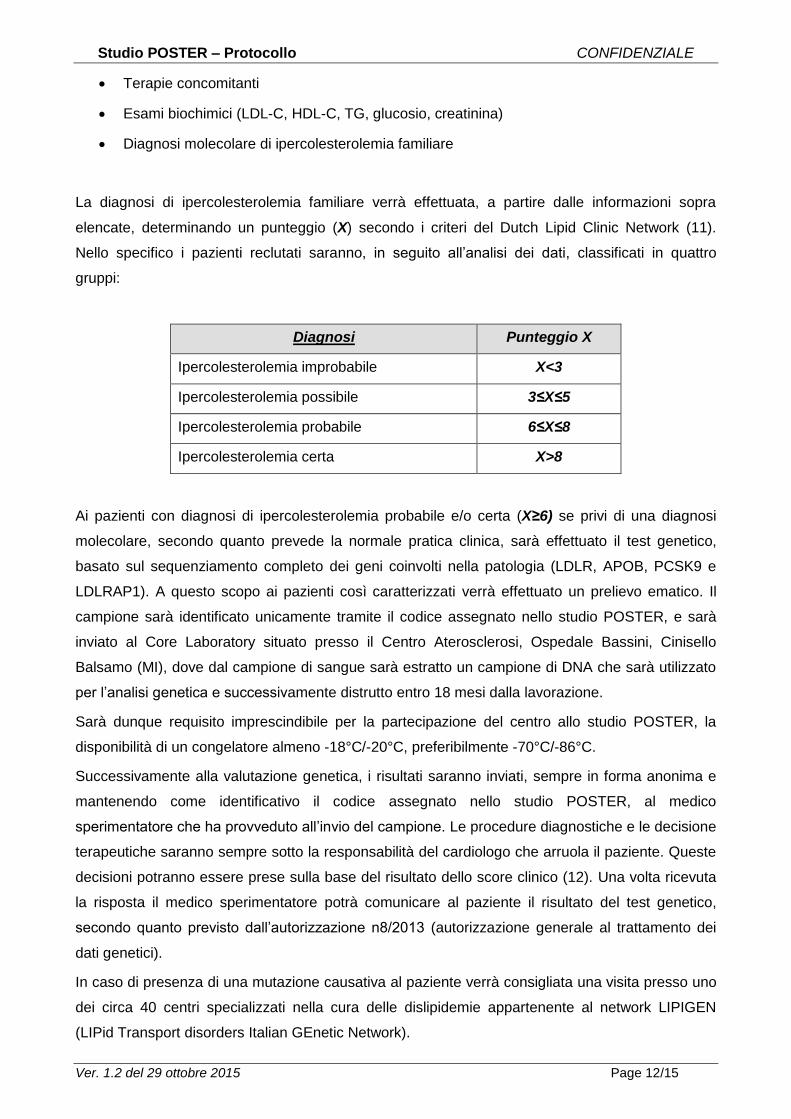
La diagnosi di ipercolesterolemia familiare verrà effettuata, a partire dalle informazioni sopra
elencate, determinando un punteggio (X) secondo i criteri del Dutch Lipid Clinic Network (11).
Nello specifico i pazienti reclutati saranno, in seguito all’analisi dei dati, classificati in quattro
gruppi:
Diagnosi Punteggio X
Ipercolesterolemia improbabile X<3
Ipercolesterolemia possibile 3≤X≤5
Ipercolesterolemia probabile 6≤X≤8
Ipercolesterolemia certa X>8
Ai pazienti con diagnosi di ipercolesterolemia probabile e/o certa (X≥6) se privi di una diagnosi
molecolare, secondo quanto prevede la normale pratica clinica, sarà effettuato il test genetico,
basato sul sequenziamento completo dei geni coinvolti nella patologia (LDLR, APOB, PCSK9 e
LDLRAP1). A questo scopo ai pazienti così caratterizzati verrà effettuato un prelievo ematico. Il
campione sarà identificato unicamente tramite il codice assegnato nello studio POSTER, e sarà
inviato al Core Laboratory situato presso il Centro Aterosclerosi, Ospedale Bassini, Cinisello
Balsamo (MI), dove dal campione di sangue sarà estratto un campione di DNA che sarà utilizzato
per l’analisi genetica e successivamente distrutto entro 18 mesi dalla lavorazione.
Sarà dunque requisito imprescindibile per la partecipazione del centro allo studio POSTER, la
disponibilità di un congelatore almeno -18°C/-20°C, preferibilmente -70°C/-86°C.
Successivamente alla valutazione genetica, i risultati saranno inviati, sempre in forma anonima e
mantenendo come identificativo il codice assegnato nello studio POSTER, al medico
sperimentatore che ha provveduto all’invio del campione. Le procedure diagnostiche e le decisione
terapeutiche saranno sempre sotto la responsabilità del cardiologo che arruola il paziente. Queste
decisioni potranno essere prese sulla base del risultato dello score clinico (12). Una volta ricevuta
la risposta il medico sperimentatore potrà comunicare al paziente il risultato del test genetico,
secondo quanto previsto dall’autorizzazione n8/2013 (autorizzazione generale al trattamento dei
dati genetici).
In caso di presenza di una mutazione causativa al paziente verrà consigliata una visita presso uno
dei circa 40 centri specializzati nella cura delle dislipidemie appartenente al network LIPIGEN
(LIPid Transport disorders Italian GEnetic Network).

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 13/15
9.3.2 Interruzione prematura dello studio
Un soggetto arruolato nello studio potrà uscire in qualsiasi momento lo desideri e per qualunque
ragione.
9.4 Gestione dei dati
Lo Sperimentatore di ogni centro o il personale da lui designato dovrà riportare le informazioni
richieste dal protocollo di studio sulla specifica Scheda Raccolta Dati (CRF) messa a disposizione
da parte dello Promotore.
I dati raccolti nella CRF saranno in forma anonima ed il soggetto verrà unicamente identificato con
un codice specifico.
Tutti i dati verranno elaborati ed analizzati dal comitato scientifico dello studio POSTER.
Tutti i dati raccolti saranno archiviati in maniera rigorosamente anonima, in base alla Linee guida
per i trattamenti di dati personali nell'ambito delle sperimentazioni cliniche di medicinali - 24 luglio
2008 G.U. n. 190 del 14 agosto 2008. I dati genetici saranno trattati secondo quanto previsto
dall’autorizzazione n8/2013 (autorizzazione generale al trattamento dei dati genetici).
L’accesso a tali dati sarà protetto dallo sperimentatore.
10 PUBBLICAZIONE DEI DATI
Non esistono vincoli alla pubblicazione dei dati da parte del promotore.
11 ACCESSO DIRETTO AI DATI ED AI DOCUMENTI ORIGINALI
A meno che non sia richiesto dalla legge, solo il medico sperimentatore od il suo staff, coinvolti
nello studio, i Comitati Etici, i Monitor, gli Study Coordinator gli Auditor e gli Ispettori rappresentanti
di organismi governativi possono accedere direttamente agli archivi per collegare i dati dello studio
ai singoli pazienti.
In casi particolari, come ad esempio durante un’ispezione da parte di agenzie regolatorie, le
informazioni legate di ogni singolo paziente e riportate in forma anonima in CRF potranno essere
collegate ai suoi dati clinici mediante la chiave conservata dallo sperimentatore.
12 ASPETTI ETICI
Lo studio dovrà essere condotto in accordo al protocollo, ai principi contenuti nella Dichiarazione di
Helsinki (ultima revisione, ottobre 2013), alle norme di Buona Pratica Clinica (ICH GCP), alle leggi
di protezione dei dati e alle altre regolamentazioni applicabili.

Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 14/15
13 REFERENZE BLIBLIOGRAFICHE
1. Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic
heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol
2004; 160(5):407-420.
2. De Castro-Oros I, Pocovi M, Civeira F. The genetic basis of familial hypercholesterolemia:
inheritance, linkage, and mutations. Appl Clin Genet 2010; 3:53-64.
3. Usifo E, Leigh SE, Whittall RA, Lench N, Taylor A, Yeats C, et al. Low-density lipoprotein
receptor gene familial hypercholesterolemia variant database: update and pathological
assessment. Ann Hum Genet 2012; 76(5):387-401.
4. Futema M, Whittall RA, Kiley A, Steel LK, Cooper JA, Badmus E, et al. Analysis of the
frequency and spectrum of mutations recognised to cause familial hypercholesterolaemia in
routine clinical practice in a UK specialist hospital lipid clinic. Atherosclerosis 2013;
229(1):161-168.
5. Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, et al. Mutations in
PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003; 34(2):154-156.
6. Garcia CK, Wilund K, Arca M, Zuliani G, Fellin R, Maioli M, et al. Autosomal recessive
hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein.
Science 2001; 292(5520):1394-1398.
7. Marks D, Thorogood M, Neil HA, Humphries SE. A review on the diagnosis, natural history,
and treatment of familial hypercholesterolaemia. Atherosclerosis 2003; 168(1):1-14.
8. Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia
in the danish general population: prevalence, coronary artery disease, and cholesterol-
lowering medication. J Clin Endocrinol Metab 2012; 97(11):3956-3964.
9. Dorsch MF, Lawrance RA, Durham NP, Hall AS. Familial hypercholesterolaemia is
underdiagnosed after AMI. BMJ 2001; 322(7278):111.
10. Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, et al. Homozygous
familial hypercholesterolaemia: new insights and guidance for clinicians to improve
detection and clinical management. A position paper from the Consensus Panel on Familial
Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J 2014;
35(32):2146-2157.
11. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS,
et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general
population: guidance for clinicians to prevent coronary heart disease: consensus statement
of the European Atherosclerosis Society. Eur Heart J 2013; 34(45):3478-3490a.
12. Catapano AL, Reiner Z, De Backer G, Graham I, Taskinen MR, Wiklund O, Agewall S,
Alegria E, Chapman M, Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Filardi PP,
Riccardi G, Storey RF, Wood D; European Society of Cardiology (ESC); European
Atherosclerosis Society (EAS). ESC/EAS Guidelines for the management of dyslipidaemias
The Task Force for the management of dyslipidaemias of the European Society of
Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis 2011;
217(1):3-46.
Studio POSTER – Protocollo CONFIDENZIALE
Ver. 1.2 del 29 ottobre 2015 Page 15/15
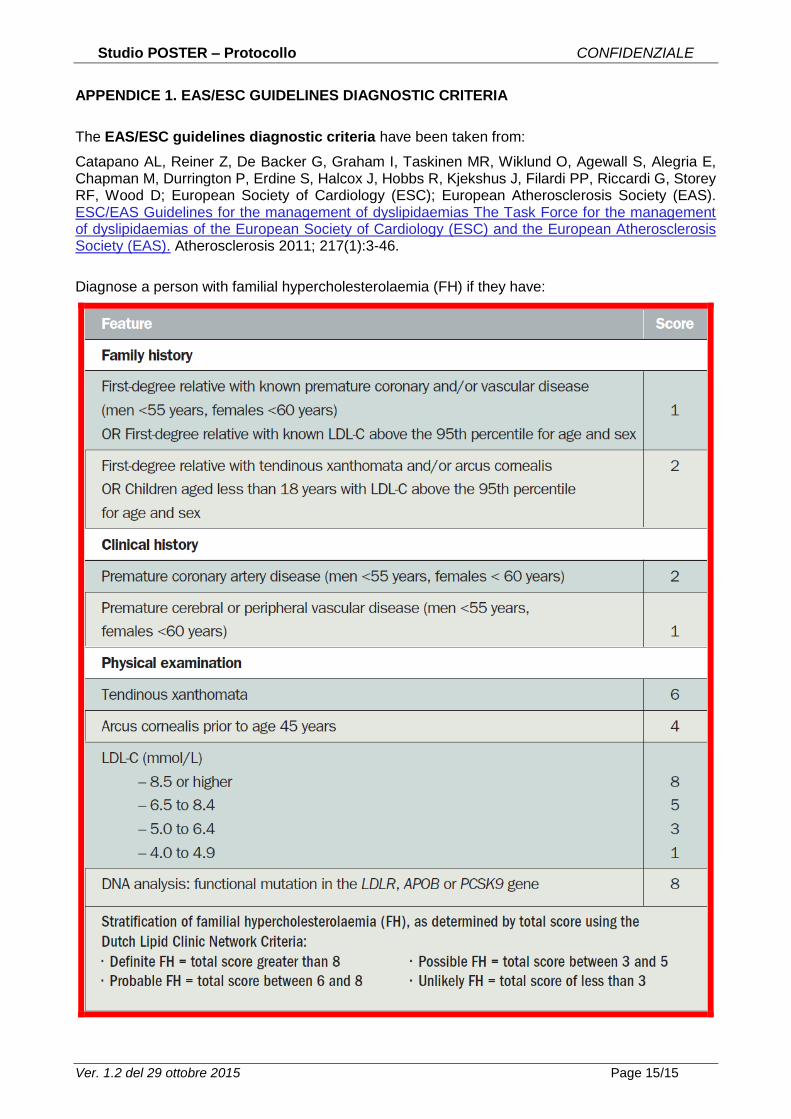
APPENDICE 1. EAS/ESC GUIDELINES DIAGNOSTIC CRITERIA
The EAS/ESC guidelines diagnostic criteria have been taken from:
Catapano AL, Reiner Z, De Backer G, Graham I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman M, Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Filardi PP, Riccardi G, Storey RF, Wood D; European Society of Cardiology (ESC); European Atherosclerosis Society (EAS). ESC/EAS Guidelines for the management of dyslipidaemias The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis 2011; 217(1):3-46.
Diagnose a person with familial hypercholesterolaemia (FH) if they have:







