Leonardo Leonardo Punzi Punzi - Luca Luca Cantarini Cantarini - Francesco Caso Francesco Caso - Paolo Paolo Sfriso Sfriso UOC di Reumatologia, AOU di Padova UOC di Reumatologia, AOU di Padova e UOC di Reumatologia, AOU Senese Siena e UOC di Reumatologia, AOU Senese Siena MALATTIE AUTOINFIAMMATORIE MALATTIE AUTOINFIAMMATORIE LIBRETTO LIBRETTO INFORMATIVO INFORMATIVO PER PAZIENTI PER PAZIENTI AFFETTI DA AFFETTI DA Con la collaborazione di Paola Galozzi
Transcript
Leonardo Leonardo PunziPunzi -- Luca Luca CantariniCantarini -- Francesco Caso Francesco Caso -- Paolo Paolo SfrisoSfriso
UOC di Reumatologia, AOU di Padova UOC di Reumatologia, AOU di Padova
e UOC di Reumatologia, AOU Senese Siena e UOC di Reumatologia, AOU Senese Siena
è probabile che lei o qualcuno della sua famiglia siate affetti daalcune manifestazioni che sono state diagnosticate dal medico comecausate da una “malattia autoinfiammatoria” .Come potrete leggere nel libretto, queste malattie, pur presenti
nell’uomo da molti anni-forse secoli o millenni-, sono state inquadratesolo di recente.
La definizione di “malattie autoinfiammatorie” si deve all’ingleseMcDermott, che ha proposto questo termine nel suo articolopubblicato nel 1999, in cui riportava la scoperta del generesponsabile della malattia TRAPS.
Il termine malattie auto-infiammatorie è stato scelto percontrapposizione alle più note e più frequenti malattie auto-immuni,caratterizzate da un difetto del sistema immunitario, conconseguente alterazione dei linfociti e produzione di auto-anticorpi.
Invece, nelle malattie auto-infiammatorie ad essere alterata è larisposta infiammatoria, un meccanismo di difesa naturale del nostroorganismo, che in questi pazienti è stimolato in maniera impropria.
In analogia con le malattie auto-immuni, anche le malattieautoinfiammatorie possono colpire vari organi e per questo vengono
In analogia con le malattie auto-immuni, anche le malattieautoinfiammatorie possono colpire vari organi e per questo vengonodette “sistemiche” (malattie autoinfiammatorie sistemiche)
In passato alcune fra le malattie autoinfiammatorie, in particolarequelle caratterizzate da febbri ricorrenti, erano classificate come“febbri periodiche ereditarie”. Il termine auto-infiammatorio èpreferibile, anche perché non tutti i pazienti presentanomanifestazioni febbrili ed inoltre, non sempre vi è dimostrazione diereditarietà.
Queste malattie insorgono soprattutto, ma non esclusivamente,nell’infanzia e la bella notizia è che, assieme alla scoperta dei geniresponsabili, sono stati identificati anche i meccanismi, portando apossibilità terapeutiche estremamente efficaci.
Purtroppo però, queste malattie risultano ancora poco conosciute,anche nell’ambito medico, con ritardi diagnostici spesso irrimediabili.
Lo scopo principale di questo libretto è di fornire ai pazienti ed ailoro parenti alcune informazioni essenziali sulle malattieautoinfiammatorie, in modo che una maggiore informazione eun’adeguata conoscenza possano migliorare la condivisione delprogramma terapeutico
Leonardo Punzi
Le malattie autoinfiammatorie sistemiche(MAIS) sono un gruppo di affezioni
caratterizzate da ricorrenti episodi di infiammazione apparentemente primitiva causati da uno sregolamento del sistema
dell’immunità innata, senza un coinvolgimento dei T-linfociti o di (auto)anticorpi specifici tipici delle
malattie autoimmuni
NON SOLO NON SOLO FEBBRE…FEBBRE…
Rispetto alla vecchia denominazione di “febbri periodiche ereditarie”,quella di malattie autoinfiammatorie sistemiche è considerata più appropriata in quanto:
a) Non tutte queste malattie sono caratterizzate da febbre
b) Alcune (forme sporadiche) non riconoscono storia familiare
c) I caratteri che accumunano maggiormente queste forme sono riferibili più ai meccanismi infiammatori che a quelli genetici
EZIOPATOGENESI EZIOPATOGENESI
La patogenesi delle malattie autoinfiammatorie è riconducibile a
mutazionimutazioni a carico di genigeni codificanti* per proteine coinvolte nella regolazione del
sistema immunitario innato, che èquello più coinvolto nella risposta
infiammatoria.
Si associano frequentemente eventi scatenanti definiti triggertrigger
che innescano la risposta infiammatoria atipica con manifestazioni sistemiche e/o
complicanze d’organo.
*I geni codificanti sono geni che portano informazioni necessarie per la sintesi di una proteina. Quindi i geni (DNA) vengono trascritti in RNA che a sua volta viene tradotto in sequenze di aminoacidi che compongono lo scheletro della proteina finale
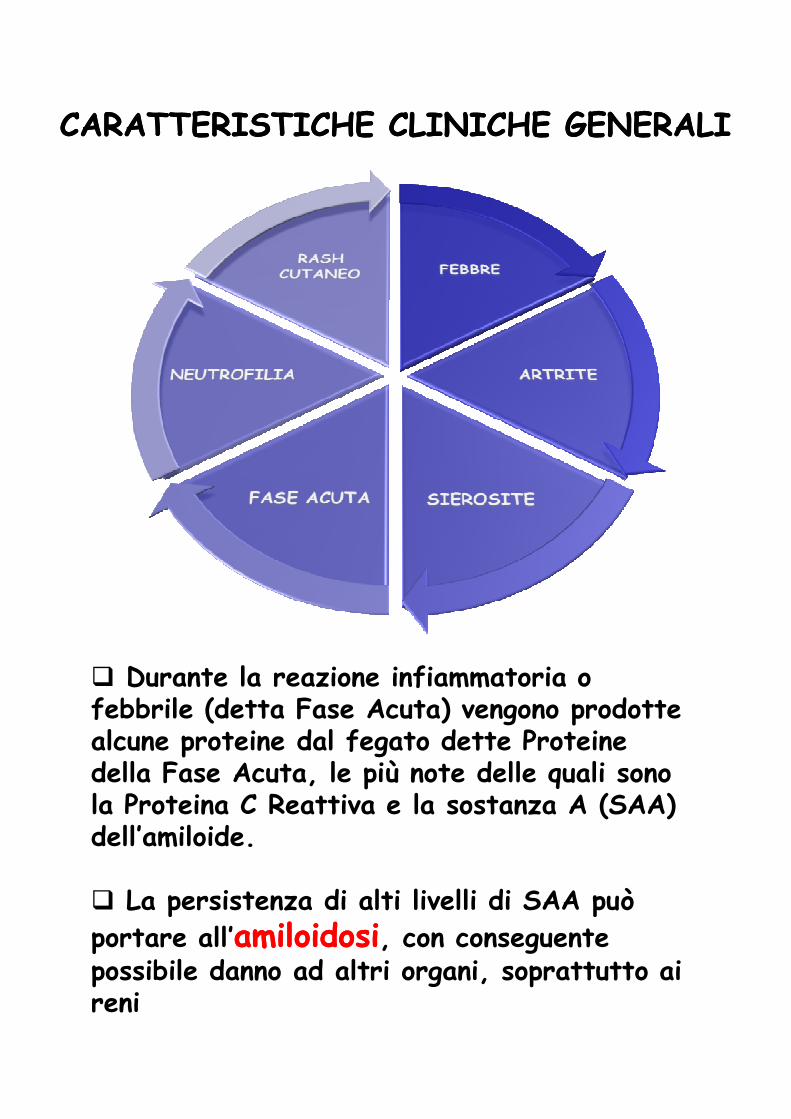
� Durante la reazione infiammatoria o febbrile (detta Fase Acuta) vengono prodotte alcune proteine dal fegato dette Proteine della Fase Acuta, le più note delle quali sono la Proteina C Reattiva e la sostanza A (SAA) dell’amiloide.
� La persistenza di alti livelli di SAA può
portare all’amiloidosiamiloidosi, con conseguente possibile danno ad altri organi, soprattutto ai reni
BREVI CENNI BREVI CENNI DIDI BIOLOGIA BIOLOGIA DELL’INFIAMMAZIONE DELL’INFIAMMAZIONE
� L’infiammazione è un meccanismo fisiologico di difesa dell’organismo, finalizzato al ripristino dei tessuti o degli organi alla loro normalità
� Può essere conseguente a cause lesive di vario tipo: fisiche (ad esempio una scottatura), chimiche (ad esempio sostanze repellenti), batteriche, immunitarie
� L’infiammazione diventa patologica (malattia) se è esagerata nell’intensità e nella durata
� L’infiammazione può essere localizzata (ad es. una scottatura) o generalizzata (ad es.febbre, artrite diffusa, pleurite). In quest’ultimo caso si definiscediffusa, pleurite). In quest’ultimo caso si definisceinfiammazione sistemica o reazione della fase acuta
� Gli elementi maggiormente responsabili dell’infiammazione sono alcune cellule (globuli bianchi) ed alcune proteine generalmente rilasciate dalle cellule e che permettono a queste di comunicare fra di loro, (citochine o interleuchine). Le più note sono il tumor necrosis factor (TNF), l’interleuchina (IL)-1 e l’IL-6
BREVI CENNI BREVI CENNI DIDI GENETICA GENETICA -- II
Glossario dei termini genetici presenti nel testo
Gene Sequenza di DNA che porta le informazioni necessarie (codifica) per la sintesi di una proteina. Rappresenta l’unità fondamentale del DNA che trasmette le informazioni genetiche da una generazione alla successiva.
- Allele una delle diverse forme che può assumere un gene.
- EsoniSegmenti di un gene che costituiscono l’ mRNA maturo, cioè il template deputato alla sintesi (codifica) della proteina.
- Introni- IntroniSegmenti genici che intervallano i diversi esoni nello stesso gene. Generalmente identificati come “materiale non codificante”, gli introni non costituiscono l’ mRNA maturo
- NucleotidiUnità base che compone la sequenza di DNA (due catene di nucleotidi avvolti a formare una spirale). Adenina (A), Citosina (C), Guanina (G) e Timina (T) sono i 4 nucleotidi.
BREVI CENNI BREVI CENNI DIDI GENETICA GENETICA -- IIII
Glossario dei termini inerenti la genetica presenti nel testo
MutazioneCambiamento nella sequenza di DNA di una cellula, stabile ereditabile e dovuto al caso o all’esposizione ai cosiddetti agenti mutageni (radiazioni o sostanze chimiche tossiche). Per convenzione, sono così considerate solo le variazioni con frequenza nella popolazione in esame < 1%.
- Si dividono in mutazioni in omozigosi quando la variazione è presente in entrambi gli alleli del gene in esame e in eterozigosi quando la variazione è presente in uno dei due alleli.- Si identificano con tecniche di sequenziamento, che - Si identificano con tecniche di sequenziamento, che consentono nel determinare l’ordine dei nucleotidi
PolimorfismoVariazioni di sequenza di DNA. Per convenzione, si considerano solo le forme con frequenza nella popolazione in esame > 1%. Si identificano anch’essi con tecniche di sequenziamento.
Penetranza
Frequenza con cui, dato un certo contesto genetico (genotipo), si manifestano i caratteri fisici (fenotipo) corrispondenti, ad esempio una malattia.
- Le mutazioni ad alta penetranza sono in grado da sole di provocare la malattia, mentre quelle a bassa penetranza conferiscono un maggior rischio di ammalarsi, ma sono incapaci da sole di provocare la malattia.
BREVI CENNI BREVI CENNI DIDI GENETICA GENETICA -- IIIIII
Glossario dei termini inerenti la genetica presenti nel testo
Malattia autosomica dominante
Causata dalla mutazione di un singolo gene, costituito da due alleli: un allele lo ereditiamo dalla madre, l'altro dal padre. Affinché la malattia si manifesti, è necessario che il figlio erediti un solo allele mutato da uno dei due genitori (definito portatore)
Malattia autosomica recessiva
Causate dalla mutazione di un singolo gene. Affinché la Causate dalla mutazione di un singolo gene. Affinché la malattia si manifesti, è necessario che il figlio erediti l'allele mutato da entrambi i genitori (definito allele recessivo).
Malattia poligenica multifattoriale
L’insorgenza dipende sia da fattori genetici (molti geni risultano coinvolti) che ambientali.
� durata di 1-3 giorni� febbre solitamente elevata (38,5-40°C)� gravità e frequenza variabile tra episodi� nell’intervallo tra gli episodi il paziente è in genere
asintomatico, tranne che per i sintomi addominali.� durante l’episodio acuto, aumentano gli indici di
infiammazione (VES, PCR)
Amiloidosirappresenta una possibile complicanza a lungo termine della FMF, legata al tipo di organo coinvolto e alla velocità di produzione e deposito di SAA a livello tessutale.
� addominali (95%)Dolore addominale, nausea, vomito, diarrea e/o stipsi. Spesso concomitanza di versamento peritoneale sterile. � articolari (75%)Artralgie (dolori articolari). Artrite (anca, ginocchio, caviglia e polso)� cutaneo (30-40%)Eritema simil-erisipeloide di diametro medio di circa 10 cm. Presenza in particolar modo sulla superficie anteriore delle gambe, tra anca e ginocchio e dorso del piede.dorso del piede.� toracico (45%)Dolore dovuto a mialgie dei muscoli della gabbiatoracica e/o a pleurite e/o pericardite.
� La diagnosi di FMF è prevalentemente clinica. � La presenza di mutazioni nel gene MEFV,
associato alla malattia, è una conferma importante, ma l’assenza non preclude la diagnosi che è posta dal medico sulla base di vari criteri, internazionalmente stabiliti
Dato il carattere di intensa infiammazione, bisogna cercare di intervenire su questa nel modo più energico possibile.
I principali farmaci disponibili a tal scopo sono:
Colchicina
Si tratta di un farmaco antico, ma tuttora efficace, estratto dalla pianta detta Colchicum autunnale. È conveniente poiché generalmente efficace e ben tollerata.
AnakinraAnakinra
Farmaco biologico che blocca i recettori di un messaggero chimico importante nell’infiammazione, l’interleuchina-1. Viene utilizzato nei casi refrattari a Colchicina.
CanakinumabFarmaco biologico specifico nel riconoscere l’interleuchina-1. Utile nei casi refrattari a Colchicina.
Altri farmaci La FMF risponde raramente al cortisone e/o ai farmaci anti-infiammatori non steroidei (FANS)
Sindrome TRAPSSindrome TRAPS
La sindrome febbrile periodica associata al recettore del TNF (TRAPS) è una malattia
genetica a trasmissione autosomicadominante, causata da mutazioni del gene
per il recettore del TNF.
� Esordio prevalentemente infantile e giovanile,ma può presentarsi anche dopo i 60 anni
Episodi febbrili ricorrenti
� Durata variabile da 1-2 giorni a diverse settimane� Frequenza di almeno 2-6 volte all’anno con risoluzione spontanea con febbre elevata (> 38.5 C)� Frequenza di almeno 2-6 volte all’anno con risoluzione spontanea con febbre elevata (> 38.5°C)� Nei periodi intercritici vi è un pieno benessere clinico, anche se nell’età adulta può venir meno l’aspetto tipico delle malattie autoinfiammatorie (caratterizzato da riaccensioni e spontanee remissioni) a favore di un decorso cronico o subcronico.
Quadro laboratoristico� Aumento del numero di globuli bianchi (leucociti) nel sangue� Aumento degli indici di infiammazione (VES, PCR, SAA, fibrinogeno e ferritina)� Aumento delle immunoglobuline sieriche (IgA e IgD)� Circa il 15-25% dei pazienti va incontro ad Amiloidosisecondaria come complicanza a lungo termine.
Sindrome TRAPSSindrome TRAPS
Altre manifestazioni
� Addominali (92%)Dolore addominale, nausea, vomito, diarrea e/o stipsi sono i sintomi d’esordio più frequenti. � Toraciche Dolore toracico e/o retrosternale in presenza di sierositi a livello pleurico e pericardico.� Articolari (75%)Artralgie periferiche, tenosinoviti, mono- o oligo-artriti a mani, ginocchia, polsi, gomiti e spalle� Cutanee Rash cutaneo eritematoso serpiginoso, con macule Rash cutaneo eritematoso serpiginoso, con macule eritematose migranti associate a dolore locale� MuscolariFascite cronica, frequenti crampi muscolari e mialgie associate ad edema e tumefazione muscolare a carico di un solo gruppo muscolare� OculariSegno caratteristico è l’edema periorbitaleassociato ad eritema monolaterale, congiutivite e dolore oculare
Sindrome TRAPSSindrome TRAPS
Diagnosi: Genetica e clinica
� Per la diagnosi di TRAPS, l’analisi genetica è determinante, in particolare lo studio degli esoni 2, 4 e 6 del gene TNFRSF1A
� Le mutazioni ad alta penetranza sonocaratterizzate da un esordio precoce della malattia e più gravi manifestazioni cliniche.
� Le mutazioni a bassa penetranza tendono ad essere associate con l'insorgenza di malattia in età adulta e presentano caratteristiche cliniche meno pronunciateE’ stato segnalato anche qualche caso di TRAPS � E’ stato segnalato anche qualche caso di TRAPS senza evidenza genetica, sulla base di osservazioni cliniche, ma tali casi necessitano ulteriori conferme
Sindrome TRAPSSindrome TRAPS
Terapia
La terapia con farmaci biologici risulta ad oggi la via migliore per i pazienti affetti da TRAPS.
� FANS e Corticosteroidi
Possono essere utili durante le fasi acute, ma non riducono la frequenza degli attacchi. Inoltre, i FANSsono scarsamente attivi e il cortisone efficace solo in una minoranza di casi a dosi medio-basse. Alcuni soggetti rispondono solo a dosi molto elevate di cortisone, improponibili per una terapia di lunga durata
� Anti Interleuchina-1Il più usato è l’anakinra, che previene l’amiloidosi e riduce la frequenza, durata e intensità di attacchi acuti. In casi particolari può essere impiegato il canakinumab, che ha una più lunga durata di azione.
� Anti TNF-αI due farmaci maggiormente impiegati sono l’etanercept e l’infliximab, che riducono l’intensità e durata degli attacchi ma con perdita di risposta nel tempo
� Anti Interleuchina-6Nei casi refrattari ad altre terapie, si può ricorrere al tocilizumab.
Deficit di Mevalonato Chinasi (MKD)Deficit di Mevalonato Chinasi (MKD)
Il deficit di Mevalonato Chinasi (MKD), definito anche Sindrome da IperIgD
(HIDS), è una malattia genetica autosomica recessiva dovuta a mutazioni del gene codificante per la mevalonato
chinasi, enzima chiave della via metabolica del colesterolo.
� La casistica maggiore proviene dal Nord Europa� L’esordio è tipicamente infantile, ma la diagnosi è possibile anche in età adulta
Episodi febbrili ricorrenti
� Durata di 3-7 giorni che ricorrono ogni 4-6 settimane con risoluzione spontanea� Febbre elevata >38,5°C� Gli attacchi acuti sembrano essere scatenati o innescati da infezioni, vaccinazioni, stress fisici ed emotivi� La frequenza degli attacchi è elevata nell’infanzia e adolescenza, ma se non adeguatamente trattata la malattia persiste in età adulta in oltre il 50% dei pazienti
Deficit da Mevalonato Chinasi (MKD)Deficit da Mevalonato Chinasi (MKD)
Quadro laboratoristico
� Aumento del numero di globuli bianchi nel sangue� Aumento degli indici di infiammazione (VES, PCR)� I livelli di IgD non sempre sono aumentati � La concentrazione urinaria di acido mevalonico può aumentare durante l’attacco acuto� L’amiloidosi è molto rara
Altre manifestazioni
� AddominaliDolori addominali, nausea, vomito, diarrea e/o Dolori addominali, nausea, vomito, diarrea e/o stipsi.� Apparato Linfatico (90%)Ingrossamento dei linfonodi (linfoadenopatia) doloroso generalizzato ed in particolare cervicale laterale. Ingrossamento della milza (splenomegalia)� ArticolariArtralgie e poliartriti non erosive simmetriche a ginocchia e caviglie� CutaneeRash maculo-papulare o più raramente orticarioide, eritematoso o purpurico� Alle mucoseAftosi ed ulcerazioni orali e genitali
Deficit da Mevalonato Chinasi (MKD)Deficit da Mevalonato Chinasi (MKD)
Genetica
La maggior parte dei pazienti MKD è eterozigote per mutazioni nel gene MVK, identificate a seguito di sequenziamento
DiagnosiLa diagnosi può essere tardiva a causa dell’aspecificitàdelle caratteristiche cliniche. L’insorgenza in età pediatrica e la ricorrenza degli attacchi indirizzano verso il sospetto diagnostico. L’analisi genetica può in seguito darne conferma.
Terapia
� FANS e Corticosteroidi
Hanno un effetto parziale nel ridurre l’intensità degli attacchi, ma possono essere sufficienti nelle forme più lievi� Farmaci anti Interleuchina-1Canakinumab e anakinra riducono la frequenza, intensità e durata degli attacchi e consentono di prevenire l’artrite.� Farmaci anti TNF-αUna buona risposta clinica è stata osservata con Etanercept.
CriopirinopatieCriopirinopatie (CAPS)(CAPS)
Gruppo di rare malattie autoinfiammatorie a trasmissione autosomica dominante, dovute a
mutazioni del gene CIAS1, noto anche come NLRP3, codificante la proteina
criopirina.
La criopirina è un’essenziale componente dell'inflammasoma, un componente dell'inflammasoma, un complesso proteico con un ruolo
primario nel controllare l’infiammazione e l’attivazione
dell’interleuchina-1 beta, citochina in grado di regolare la risposta
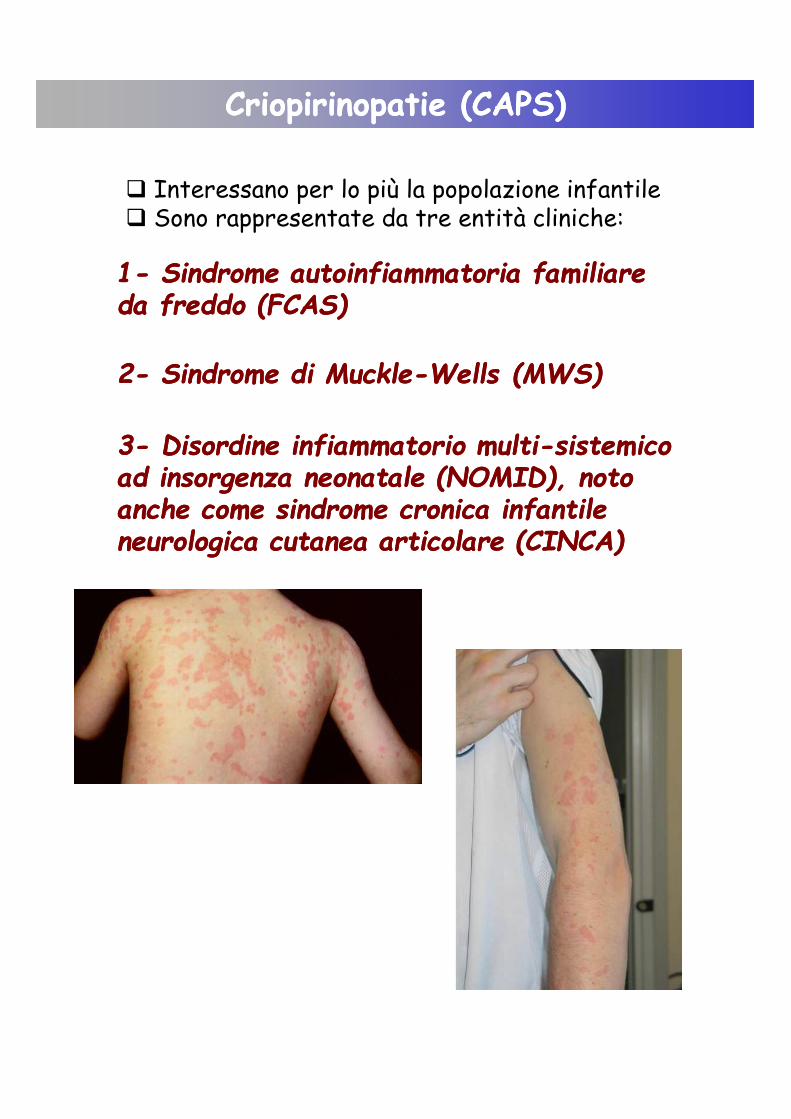
33-- Disordine infiammatorio Disordine infiammatorio multimulti--sistemicosistemicoad insorgenza neonatalead insorgenza neonatale (NOMID), noto (NOMID), noto anche come sindrome cronica infantile anche come sindrome cronica infantile neurologica cutanea articolare (CINCA)neurologica cutanea articolare (CINCA)
� Interessano per lo più la popolazione infantile� Sono rappresentate da tre entità cliniche:
CriopirinopatieCriopirinopatie
11-- Sindrome Sindrome autoinfiammatoriaautoinfiammatoria familiare familiare da da freddo (FCAS)freddo (FCAS)
� Descritta per la prima volta nel 1940 � Esordio nei primi mesi di vita
Episodi febbrili ricorrenti
� Breve durata degli attacchi (1-2 ore fino ad un massimo di 24 ore) con risoluzione spontanea� I sintomi solitamente compaiono in seguito all’esposizione alle basse temperature o a cambiamenti rapidi di temperatura, con peggioramenti serali.
Quadro laboratoristico� Aumento degli indici di infiammazione (VES, PCR, e fibrinogeno) durante gli episodi acuti� Non si associa ad amiloidosi e sordità, a differenza delle altre criopirinopatie
Altre manifestazioni� ArticolariStanchezza (astenia), artralgie e/o artriti associate a rigidità articolare.� CutaneeRash cutaneo papulare simil-orticarioide migrante.� Oculari Infiammazione oculare con congiuntivite.
CriopirinopatieCriopirinopatie
22-- Sindrome di Sindrome di MuckleMuckle--WellsWells (MWS)(MWS)
� Esordio in età infantile (nei primi mesi di vita)� Può non presentarsi con febbre
Episodi febbrili ricorrenti
� Attacchi acuti di breve durata (massimo 24 ore) in seguito ad esposizione a basse temperature o rapidi cambi climatici, con peggioramento serale � Possono essere favoriti da stress fisico, emotivo e processi infettivi
Quadro laboratoristico� Aumento degli indici di infiammazione (VES, PCR) � Aumento degli indici di infiammazione (VES, PCR) � Nel 25% dei pazienti si sviluppa amiloidosi
Altre manifestazioni� CutaneeLesioni simil-orticarioidi migranti ed evanescenti� OculariCongiuntivite, atrofia del nervo ottico che può portare alla cecità� ArticolariArtralgie e/o poliartrite non erosiva� Uditive Alla malattia si associa sordità neuro-sensoriale nel 70% dei pazienti con insorgenza sia durante l’infanzia che in età adulta.
� Rappresenta il fenotipo più grave delle CAPS� Esordio fin dalle prime settimane di vita
Quadro laboratoristico
� Leucocitosi, anemia cronica� Aumento dei livelli sierici di proteine della fase acuta e VES
Altre manifestazioni� Cutanee (75%)� Cutanee (75%)Rash cutaneo cronico simil-orticarioide, non pruriginoso
� Oculari Uveite anteriore cronica, atrofia del nervo ottico con riduzione della vista fino alla cecità
� Muscolo-scheletricheAtropatia ipertrofica a carico di mani, ginocchia e piedi, con prematura ossificazione; poliartrite cronica erosiva; dismorfismo del viso
� Sistema nervoso centraleMeningite asettica cronica, atrofia celebrale
CriopirinopatieCriopirinopatie (CAPS)(CAPS)
Genetica
� FCAS e Sindrome di Muckle-Wells hanno carattere familiare mentre NOMID è sporadica.� In circa il 30% dei pazienti non si riscontrano mutazioni nel gene associato NLRP3 (o CIAS1)
Terapia
I farmaci che bloccano l’interleuchina-1 sono in grado di controllare i sintomi acuti e prevenire gli episodi febbrili nelle tre forme CAPS.
Canakinumab� Canakinumab
Farmaco biologico specifico nel riconoscere l’interleuchina-1 beta. Controlla sin dalle prime somministrazioni i sintomi acuti e riduce la frequenza degli episodi nelle tre forme. È inoltre efficace nel migliorare l’udito in NOMID� Anakinra
Farmaco biologico specifico nel riconoscere l’interleuchina-1 beta. Controlla sin dalle prime somministrazioni i sintomi acuti e riduce la frequenza degli episodi nelle tre forme.
Sindrome di Blau
La Sindrome di Blau è una rara malattia granulomatosa autosomica dominante,
caratterizzata principalmente da artrite, rash cutaneo e uveite.
� Esordio nella prima infanzia e solitamente in età superiori ai 5 anni di vita.� All’esordio, risulta caratterizzata da sintomi articolari e cutanei; i sintomi oculari sono più tardivi e compaiono tra i 7 e i 12 anni.
Quadro clinico
� Artrite granulomatosa
Sindrome di BlauSindrome di Blau
� Artrite granulomatosaPrevalentemente poliarticolare simmetrica, a carico di polsi, mani, caviglie, piedi e gomiti.
� Rash cutaneo papulo-nodulare color rosso-marrone, intermittente e con possibile esito cicatriziale.
� Manifestazioni oculariGravi, con uveite anteriore o panuveite granulomatosa. L’infiammazione può interessare qualsiasi struttura oculare, determinando cataratta, glaucoma, distacco della retina e talvolta cecità completa.
� Altre manifestazioniFebbre persistente o intermittente; insufficienza epatica e renale; interessamento del sistema nervoso.
Sindrome di Blau
Genetica � Il sequenziamento dell’esone 4 consente di
individuare le maggiori mutazioni del gene CARD15/NOD2 associate alla Sindrome di Blau.
Terapia
� Non vi è ancora un trattamento ottimale per i pazienti con sindrome di Blau� Nella fase quiescente di malattia, basse dosi di cortisonici sono efficaci, mentre in fase acuta vanno considerati più alti dosaggi.
Sindrome di BlauSindrome di Blau
vanno considerati più alti dosaggi.� In caso di inefficacia dei glucorticoidi, i farmaci biologici (infliximab, anakinra) possono rappresentare una valida alternativa.
Sindrome DIRA Sindrome DIRA
La malattia da deficit dell’antagonista recettoriale dell’interleuchina 1 (DIRA)
è una malattia a trasmissione autosomica recessiva, causata da
mutazioni nel gene IL1RN.
� Esordio già dal primo anno di vita
Quadro clinico e laboratoristico� Caratterizzata da un processo infettivo che interessa simultaneamente le ossa ed il midollo osseo (osteomielite) e da pustolosi.� Coinvolgimento osseo caratterizzato da lesioni osteolitiche e rigonfiamento delle due estremità osteolitiche e rigonfiamento delle due estremità tondeggianti (epifisi) delle ossa lunghe.� Coinvolgimento cutaneo con lesioni pustolose di dimensioni variabili� Indici di infiammazione persistentemente elevati
Genetica
� Il gene associato alla malattia (IL1RN) esprime la proteina “antagonista del recettore dell’interleuchina 1”, inibitore dell’attività dell’interleuchina 1 nel legare il suo specifico recettore IL1R
Terapia� Efficace l’uso del farmaco biologico anti interleuchina-1 (Anakinra)
Sindrome di Sindrome di MajeedMajeed
La Sindroma di Majeed è una rara malattia a trasmissione autosomicarecessiva causata da mutazioni del
gene LPIN2.
� Esordio nei primi 2 anni di vita
Quadro clinico
� Episodi acuti ricorrenti di breve durata con altrettanto brevi fasi di remissione� Osteomielite sterile cronica ricorrente multifocale� Anemia� Anemia� Dermatosi infiammatoria neutrofilica
Terapia
� Corticosteroidi
� FANS
Genetica
� Il gene associato alla malattia (LPIN22) esprime la proteina “Phosphatidate phosphataseLPIN2”, che gioca un ruolo importante nel controllare il metabolismo degli acidi grassi.
Sindrome DITRA Sindrome DITRA
Il Deficit dell’antagonista recettoriale dell’interleuchina 36 (DITRA) è una malattia a trasmissione autosomica recessiva, causata da mutazioni del
gene IL36RN
� Esordio in età infantile
Quadro clinico e laboratoristico� Episodi acuti febbrili ricorrenti� Rash eritematoso e pustoloso� Astenia� Leucocitosi ed aumento degli indici di infiammazione
Genetica
� Il gene associato alla malattia (IL36RN) esprime la proteina “antagonista del recettore dell’inteleuchina36”, inibitore dell’attivazione di NF-κB, componentechiave dei processi infiammatori cellulari
Terapia� Corticosteroidi
� Immunosoppressori
� Farmaci biologici anti TNF-α� Acitretina
Farmaco ampiamente utilizzato nel trattamento delle forme gravi di psoriasi, combatte la proliferazione delle cellule
Sindrome PAPASindrome PAPA
L'artrite piogenica sterile (pioderma gangrenoso) con acne (Sindrome
PAPA) è una malattia a trasmissione autosomica dominante associata a
mutazioni del gene PSTPIP1.
� Esordio nella prima infanzia
Quadro clinico
� Artrite piogenica sterile che può coinvolgere da una a tre articolazioni ed avere risoluzione spontanea� Manifestazioni cutanee:
Terapia
� Corticosteroidi� Farmaci biologici anti interleuchina-1 (Anakinra) e TNF-α
Genetica
� Il gene associato alla malattia (PSTPIP1) esprime la proteina “CD2 antigen-binding protein 1”, che ha il compito principale di inibire i segnali infiammatori mediati dalla proteina pirina (legata alla Febbre Mediterranea Familiare).
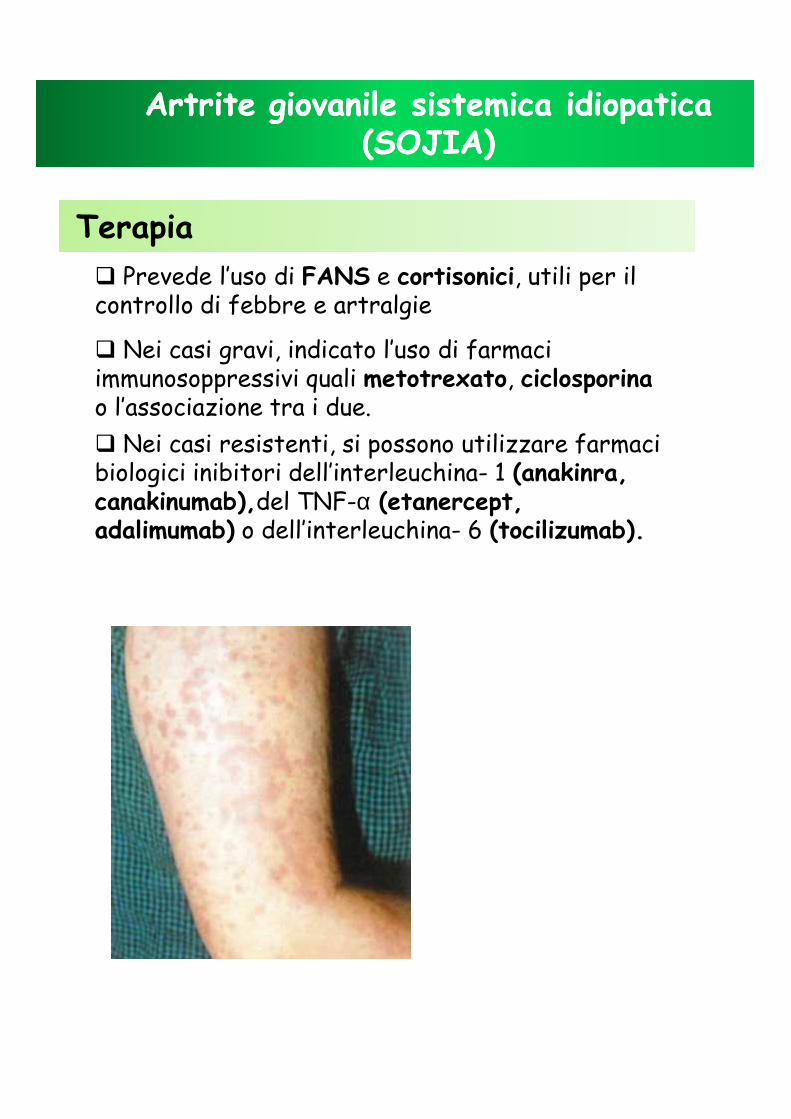
� Manifestazioni cutanee:pioderma gangrenoso, lesioni ulcerative a livello degli arti inferiori, acne cistica.
Sindrome di BehçetSindrome di Behçet
La sindrome di Behçet è una malattia autoinfiammatoria sistemica cronica, a eziologia sconosciuta, caratterizzata da
aftosi orale e genitale ricorrente, artrite e infiammazione oculare
� Prevalenza nei territori lungo l’antica Via della Seta, con un’incidenza di 7.5 soggetti affetti su 100 000 in Europa.� Esordio raro dopo i 55 anni � Prevalenza nei maschi delle forme più sintomatiche
Quadro clinico� Andamento temporale (episodi ricorrenti e periodi di � Andamento temporale (episodi ricorrenti e periodi di remissione spontanea)� Infiammazione sistemica� Possibili associazioni con polimorfismi dei geni MEFV eTNFRSF1A, caratteristici della FMF e della TRAPS.
Quadro laboratoristico� Nota l’associazione con HLA-B51 � Assenza di iper-reattività delle cellule B o T� Aumento degli indici di infiammazione e medio incremento di globuli bianchi� Incremento delle IgG e IgM
Sindrome di BehçetSindrome di Behçet
Altre manifestazioni� Manifestazioni mucocutanee
Afte orali (98%) e genitali (60-65%) dolorose, isolate o raggruppate, che guariscono in 1-2 settimane senza lasciare esiti cicatriziali� Manifestazioni oculari (70%)� Manifestazioni articolari (50%)
� Manifestazioni vascolari
La vasculite è l’aspetto istologico principale. � Manifestazioni neurologiche� Manifestazioni polmonari� Manifestazioni gastrointestinali� Manifestazioni linfatiche� Manifestazioni linfatiche
Diagnosi� Criterio essenziale per la diagnosi è l’aftosi orale ricorrente, associata alla presenza di almeno due manifestazioni cutanee e/o oculari
TerapiaIl trattamento varia a seconda dell’estensione e della gravità della forma.� Corticosteroidi come trattamento cardine� Farmaci immunosoppressori di affiancamento� Nei casi più severi, farmaci biologici anti-TNF e anti-interleuchina 1
� Colchicina efficace per le lesioni mucocutanee
La Febbre periodica con stomatite aftosa, faringite, adenite cervicale (PFAPA) è una
malattia tipica dell’età pediatrica, caratterizzata da episodi febbrili
ricorrenti e stomatiti aftose
� Esordio tipico dell’età pediatrica, al di sotto dei 5 anni.� Ruolo determinante nell’insorgenza lo rivestono i fattori ambientali e la predisposizione genetica.
Quadro clinico
� Episodi febbrili (>38,5-40°C) con insorgenza ogni 4-6 settimane e periodi tra gli episodi variabili e caratterizzati da sintomaticità.
Sindrome PFAPASindrome PFAPA
caratterizzati da sintomaticità.� Stomatite aftosa e faringite� Ingrossamento dei linfonodi (linfoadenopatia) a livello cervicale
Genetica
� Variazioni nel gene PSTPIP2 sono state associate alla malattia
La diagnosi è prevalentemente di esclusione di altre patologie quali le immunodeficienze primarie e le altre febbri periodiche.
Terapia
Basse dosi di corticosteroidi durante le fasi acute
Diagnosi
Morbo di Morbo di StillStill dell’adulto (AOSD)dell’adulto (AOSD)
Il Morbo di Still dell’adulto o Adult Onset Still Disease (AOSD) è una
malattia autoinfiammatoria multifattoriale rara di origine sconosciuta,
caratterizzata da alti picchi febbrili, artriti, rash cutaneo e aumento del numero di globuli bianchi nel sangue
(leucocitosi neutrofila).
� Malattia rara con incidenza 0.16 per 100.000 abitanti� Picchi di insorgenza a 15-25 anni e 36-46 anni
Quadro clinico
� Febbre transitoria di durata < 4ore� 1-2 picchi febbrili >39°C giornalieri � Rash cutaneo Maculo-papulare, evanescente e talvolta pruriginoso a livello dell’estremità prossimale di arti, tronco. L’andamento segue gli episodi febbrili.� Artralgie ed artriti
Coinvolgimento oligo/poliarticolare, solitamente bilaterale, ed interessamento di ginocchia, polsi e caviglie.� Mialgie
� Sierositi (pleuriti e pericarditi)� Ingrossamento dei linfonodi e della milza (linfo/splenomegalia)
Morbo di Morbo di StillStill dell’adulto (AOSD)dell’adulto (AOSD)
Quadro laboratoristico
� Marcato aumento dei globuli bianchi (leucocitosi) e delle piastrine (trombocitosi)� Aumento degli indici di infiammazione (VES, PCR, SAA e fibrinogeno)� L’aumento dell’SAA può causare amiloidosisecondaria
DiagnosiI sintomi e i segni dell’AOSD possono confondersi con quelli di altre malattie, anche gravi. Per questo è necessaria la valutazione reumatologica il più precocemente possibile. Esitono dei criteri precocemente possibile. Esitono dei criteri internazionali per inquadrare opportunamente questi pazienti
Terapia� Corticosteroidi ad alti dosaggi
� Metotrexato di fondo� Anti interleuchina-1 Anakinra e Canakinumab controllano fin dalle prime somministrazioni il rash cutaneo, la febbre e l’artrite nei casi refrattari ad altre terapie� Anti interleuchina-6Tocilizumab controlla l’intensità e la gravità delle manifestazioni artro-cutanee e previene le recidive nei casi refrattari ad altre terapie
Le artriti idiopatiche giovani sono un gruppo eterogeneo di malattie articolari
infiammatorie ad eziologia sconosciuta che insorgono prima del 16° anno d’età.
La forma sistemica è nota anche come Morbo di Still
� Malattia rara, con frequenza di 4-17% tra tutte le artriti giovanili idiopatiche� Insorge entro i 16 anni, con un picco attorno ai 2 anni e un rapporto 1:1 tra maschi e femmine
Quadro clinicoQuadro clinico
� Febbre elevata della durata di almeno 2 settimane con picchi >39°C e quotidiana per almeno 3 giorni consecutivi.� Rash cutaneo Maculo-papulare, evanescente e talvolta pruriginoso a livello dell’estremità prossimale di arti, tronco. � Artralgie ed artritiCoinvolgimento poliarticolare, solitamente bilaterale, delle grandi e piccole articolazioni degli arti, la colonna cervicale e le articolazioni temporo-mandibolari.� Sierositi (pleuriti e pericarditi)
� Ingrossamento dei linfonodi e della milza
� Rare complicanze: Oculari, ritardo della crescita
Quadro laboratoristico� Marcato incremento di globuli bianchi (fino a 20-50.000 cellule/mm3)� Aumento degli indici di flogosi (VES, PCR), piastrinosi e anemia� Fattore reumatoide e anticorpi anti-nucleo ANA generalmente negativi� Aumento elevato della ferritina sierica
Diagnosi
� L’esclusione dei criteri sotto elencati portano alla � L’esclusione dei criteri sotto elencati portano alla certezza di diagnosi.
Psoriasi o storia familiare di psoriasi in un parente di I grado
� Prevede l’uso di FANS e cortisonici, utili per il controllo di febbre e artralgie
� Nei casi gravi, indicato l’uso di farmaci immunosoppressivi quali metotrexato, ciclosporinao l’associazione tra i due.� Nei casi resistenti, si possono utilizzare farmaci biologici inibitori dell’interleuchina- 1 (anakinra, canakinumab),del TNF-α (etanercept, adalimumab) o dell’interleuchina- 6 (tocilizumab).